
سندرم نونان
Noonan Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، مهسا جمالی (دانشجوی کارشناسی ارشد ژنتیک), رویا آزاد خواه (دانشجوی کارشناسی ارشد ژنتیک) ، دکتر نعیمه شیبایی (دکترای تخصصی ژنتیک مولکولی)، سهیل باغبان (دانشجوی کارشناسی ارشد ژنتیک)، مجید عجم زاده (دانشجوی کارشناسی ارشد ژنتیک)
نگارنده مسئول: شاهین اسعدی (Molecular Geneticist)

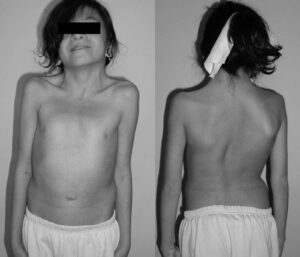
سندرم نونان یک اختلال ژنتیکی است که علائم آن بهطورمعمول در هنگام تولد، آشکار میشود. این اختلال توسط طیف گستردهای از علائم فیزیکی بر اساس محدوده و شدت بیماری، قابلتشخیص است. در بسیاری از افراد مبتلا به این سندرم، ناهنجاریهایی مانند: ظاهر متمایز صورت، گردن گسترده و یا پردهدار مشهود است و همچنین بدشکلی قفسه سینه و کوتاهی قد از دیگر ویژگیهای سندرم نونان میباشد. بعلاوه ناهنجاریهایی مانند: انحنای ستون فقرات (اسکولیوز)، نقص ماهیچههای قلب، تنگی دریچه ریوی و ضخیم شدن عضله بطن قلب (هایپرتروفیک کاردیومیوپاتی) نیز در افراد مبتلا به سندرم نونان مشاهده میشود.
اختلالات دیگر ممکن است شامل: ناهنجاریهای خون و عروق لنفاوی خاص، لخته شدن خون و کمبود پلاکت، مشکلات یادگیری و یا ناتوانی فکری خفیف، شکست لایه سطحی بیضهها و فرود به داخل کیسه بیضه طوریکه به ظاهر، فرد مبتلا به این سندرم فقط دارای یک بیضه است، در سال اول زندگی مردان مبتلا به سندرم نونان، مشاهده گردد.
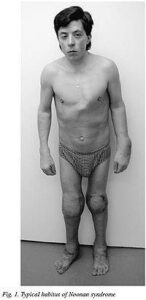
علائم و نشانههای بالینی
اکثر شیرخواران مبتلا به این سندرم دارای ویژگیهای سروصورت مشخص هستند. در بسیاری از موارد، سر نسبتاً بزرگ به نظر میرسد. نوزادان مبتلا ممکن است دارای افتادگی پلک بالا، شکاف پلک، چینهای پوست در اطراف چشم و رنگ چشم کاملاً آبی یا مایل به آبی باشند. بسیاری از نوزادان با سندرم نونان، وزنشان در هنگام تولد طبیعی است. بااینحال در برخی از نوزادان، هنگام تولد ممکن است به دلیل تجمع غیرطبیعی مایع بین لایههای بافت زیرپوست (ادم زیر جلدی)، وزنشان افزایش یابد. بهعنوانمثال؛ تورم پشت دست و بالای پا در نوزادان با این سندرم مشترک است. برخی از نوزادان مبتلا به سندرم نونان، ممکن است مشکلات تغذیهای را تجربه کنند و با اختلال رشد همراه باشند. ارتفاع متوسط قامت مردان بالغ مبتلا به سندرم نونان، 162.5 سانتیمتر و ارتفاع متوسط قامت زنان بالغ مبتلا به این سندرم، 152.7 سانتیمتر است.

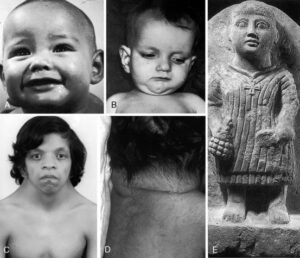
برخی مردان و زنان مبتلا به این سندرم، نیز ممکن است اختلال در توسعه ویژگیهای ثانویه جنسی را تجربه کنند. در حدود 60 تا 75 درصد از مردان مبتلا به سندرم نونان، یک یا هر دو لایه سطحی بیضه به داخل کیسه بیضه فرو رفته است که این فرایند قبل از تولد یا در طول سال اول زندگی رخ میدهد. در چنین مواردی اگر جراحی جهت تصحیح فرم بیضهها، صورت نگیرد، ممکن است مردان مبتلا به این سندرم دچار کمبود سلولهای اسپرماتوژنز شده و عقیمی را تجربه کنند؛ اما بسیاری از زنان با سندرم نونان، ازنظر باروری، طبیعی هستند.
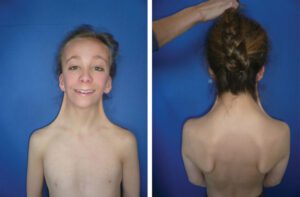
تقریباً %70 نوزادان مبتلا به سندرم، در هنگام تولد، اختلالات قلبی عروقی دارند و در حدود نیمی از این موارد، نوزادان گرفتار انسداد جریان طبیعی خون از پائین بطن راست قلب به ریهها (تنگی عروق ریوی) میباشند. حدود %33-20 از افراد مبتلا به این سندرم، نقصهای مختلف لخته شدن خون (کمبود فاکتور انعقادی)، سطوح پائین گردش پلاکتها در خون (ترومبوسیتوپنی) و یا عملکرد نادرست از پلاکتهای خون را دارند.
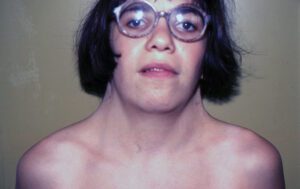
برخی از افراد مبتلا به این سندرم، ممکن است تغییر در رنگ پوست داشته باشند. در حدود %25 از افراد مبتلا ممکن است خال زیاد در پوست وجود داشته باشد. در %35 از افراد مبتلا به سندرم نونان ممکن است ناتوانی فکری خفیف وجود داشته باشد. همچنین تأخیر در یادگیری، هماهنگی عضلات در حین انجام کار، تأخیر در زبان گفتاری و از دست دادن حس شنوایی خفیف در مبتلایان این سندرم مشهود است.


ژنتیک مولکولی
سندرم نونان از الگوی توارثی اتوزومال غالب پیروی میکند. تا به حال 18 ژن برای ایجاد این سندرم، ( اعم از حیوانات و انسان ) شناسایی شدند که 14 ژن از آن، در انسان سندرم نونان را ایجاد میکند و از این میان، 5 ژن در اغلب موارد سندرم نونان مشهود است. شایعترین ژن جهش یافته در %50 از موارد سندرم نونان، جهش در ژن PTPN11 میباشد.
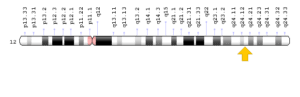
A ) ژن PTPN11 که در بازوی بلند کروموزوم شماره 12 بهصورت 12q24.13 مستقر است.

B ) جهش در ژن SOS1 که در بازوی کوتاه کروموزوم شماره 2 بهصورت 2p22.1 مستقر است و %13-10 از موارد این سندرم را ایجاد میکند.
 C ) جهش در ژن RAF1 که در بازوی کوتاه کروموزوم شماره 3 بهصورت 3p25.2 مستقر است و %5 از موارد این سندرم را ایجاد میکند.
C ) جهش در ژن RAF1 که در بازوی کوتاه کروموزوم شماره 3 بهصورت 3p25.2 مستقر است و %5 از موارد این سندرم را ایجاد میکند.
 D ) جهش در ژن RIT1 که در بازوی بلند کروموزوم شماره 1 بهصورت 1q22 مستقر است و %5 از موارد این سندرم را ایجاد میکند.
D ) جهش در ژن RIT1 که در بازوی بلند کروموزوم شماره 1 بهصورت 1q22 مستقر است و %5 از موارد این سندرم را ایجاد میکند.
 E ) جهش در ژن KRAS که در بازوی کوتاه کروموزوم شماره 12 بهصورت 12p12.1 مستقر است و کمتر از %5 موارد این سندرم را ایجاد میکند.
E ) جهش در ژن KRAS که در بازوی کوتاه کروموزوم شماره 12 بهصورت 12p12.1 مستقر است و کمتر از %5 موارد این سندرم را ایجاد میکند.
 F ) جهش در ژن SOS2 که در بازوی بلند کروموزوم شماره 14 بهصورت 14q21.3 مستقر است و %1 موارد این سندرم را ایجاد میکند.
F ) جهش در ژن SOS2 که در بازوی بلند کروموزوم شماره 14 بهصورت 14q21.3 مستقر است و %1 موارد این سندرم را ایجاد میکند.
 G ) جهش در ژن A2ML1 که در بازوی کوتاه کروموزوم شماره 12 بهصورت 12p13.31 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
G ) جهش در ژن A2ML1 که در بازوی کوتاه کروموزوم شماره 12 بهصورت 12p13.31 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
 H ) جهش در ژن RASA2 که در بازوی بلند کروموزوم شماره 3 بهصورت 3q23 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
H ) جهش در ژن RASA2 که در بازوی بلند کروموزوم شماره 3 بهصورت 3q23 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
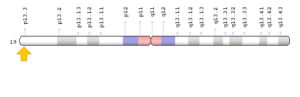 J ) جهش در ژن MEK2 که در بازوی کوتاه کروموزوم شماره 19 بهصورت 19p13.3 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
J ) جهش در ژن MEK2 که در بازوی کوتاه کروموزوم شماره 19 بهصورت 19p13.3 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
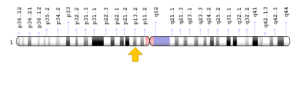
K ) جهش در ژن NRAS که در بازوی کوتاه کروموزوم شماره 1 بهصورت 1p13.2 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
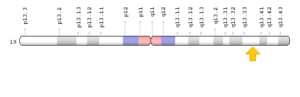
M ) جهش در ژن RRAS که در بازوی بلند کروموزوم شماره 19 بهصورت 19q13.40 مستقر است و %1 از موارد این سندرم را ایجاد میکند.
 N ) جهش در ژن BRAF که در بازوی بلند کروموزوم شماره 7 بهصورت 7q34 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.
N ) جهش در ژن BRAF که در بازوی بلند کروموزوم شماره 7 بهصورت 7q34 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.
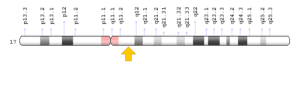 S ) جهش در ژن LZTR1 که در بازوی بلند کروموزوم شماره 22 بهصورت 22q11.21;22q11.1-q11.2 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.
S ) جهش در ژن LZTR1 که در بازوی بلند کروموزوم شماره 22 بهصورت 22q11.21;22q11.1-q11.2 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.

R ) جهش در ژن SHOC2 که در بازوی بلند کروموزوم شماره 10 بهصورت 10q25 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.

T ) جهش در ژن CBL که در بازوی بلند کروموزوم شماره 11 بهصورت 11q23.3 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.
L ) جهش در ژن NF1 که در بازوی بلند کروموزوم شماره 17 بهصورت 17q11.2 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.
P ) جهش در ژن MAP2K2 که در بازوی کوتاه کروموزوم شماره 19 بهصورت 19p13.3 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.
Q ) جهش در ژن MAP2K1 که در بازوی بلند کروموزوم شماره 15 بهصورت 15q22.31 مستقر است و %1 از موارد سندرم نونان را ایجاد میکند.
فرکانس
سندرم نونان، مردان و زنان را به یک میزان تحت تأثیر قرار میدهد و فراوانی این سندرم تقریباً 1 در 1000 یا 1 در 2500 تولد زنده میان همه نژادهای انسانی میباشد. این بیماری یکی از شایعترین سندرمهای ژنتیکی در بروز بیماریهای قلبی عروقی مادرزادی، مشابه فرکانس سندرم داون میباشد.


تشخیص سندرم
در برخی موارد سندرم نونان ممکن است، قبل از تولد نوزاد از طریق سونوگرافی بر اساس مشاهده گردن نوزاد که بهصورت پردهدار میباشد، تشخیص داده شود. بااینحال قطعیترین تست جهت تشخیص سندرم نونان، آزمایش ژنتیک مولکولی در ژنهای کاندید برای سندرم نونان میباشد.
مسیرهای درمانی سندرم
در حال حاضر هیچ درمان قاطعی برای سندرم نونان وجود ندارد، اما میتوان با روشهای نوین پزشکی به رنج بیماران، تخفیف داد. برای مثال: در بیمارانی که مبتلا به اختلالات قلبی عروقی هستند و تنگی دریچه ریوی دارند، میتوان با عمل جراحی این اختلال را تا حدودی برطرف کرد. همچنین مبتلایانی که در فاکتورهای انعقادی خون خود دچار نقص هستند، میتوان با تجویز فاکتورهای انعقادی و پلاکت از رنج بیماران کاست. بعلاوه برای جلوگیری از خطر عقیمی مردان مبتلا به سندرم نونان با فرورفتگی لایه سطحی بیضهها در کیسه بیضه، عمل جراحی انجام داد. فیزیوتراپی برای درمان اختلالات عضلانی بیماران و گفتاردرمانی برای بهبود کیفیت تکلم بیماران سندرم نونان، مؤثر میباشد. درنهایت، مشاوره ژنتیک قبل از بارداری بهمنظور جلوگیری یا کاهش تولد نوزادان سندرم نونان برای والدین نیز امری ضروری میباشد.
تاریخچه سندرم
سندرم نونان اولین بار در سال 1883 توسط دکتر اسکار کوبیلینسکی گزارش گردید، اما بهطور کاملتر توسط دکتر (J.A. Noonan and D.A. Ehmke) در سال 1963 گزارش گردید.

تصویر دکتر نونان، کاشف سندرم
References:
- 1. James, William; Berger, Timothy; Elston, Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders.
- Curcić-Stojković O, Nikolić L, Obradović D, Krstić A, Radić A (1978). “[Noonan’s syndrome. (Male Turner’s syndrome, Turner-like syndrome)]”. Med Pregl. 31 (7–8): 299–303.
- 3. Imbalance of plasminogen activator inhibitor type-1 (PAI-1) and tissue plasminogen activator (t-PA) activity in patients with Noonan syndrome.”. J Pediatr Hematol Oncol. 32 (7): 532–6. Oct 2010.
- 4. Reinker, Kent; Stevenson DA; Tsung A (July–August 2011). “Orthopaedic conditions in Ras/MAPK related disorders.”. Journal of Pediatric Orthopeadics. 31 (5): 599–605.
- 5. Growth hormone and noonan syndrome: update in dysfunctional signaling aspects and in therapy for short stature”. Hormonal Studies. 2: 1.
- Razzaque MA, Komoike Y, Nishizawa T, et al. (March 2012). “Characterization of a novel KRAS mutation identified in Noonan syndrome”. Am. J. Med. Genet. A. 158A (3): 524–32.
- Tartaglia M, Mehler EL, Goldberg R, et al. (2001). “Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome”. Nat. Genet. 29 (4): 465–8.
- Shchelochkov OA, Patel A, Weissenberger GM, et al. (April 2008). “Duplication of chromosome band 12q24.11q24.23 results in apparent Noonan syndrome”. Am. J. Med. Genet. A. 146A (8): 1042–8.
- Van Der Burgt, I.; Brunner, H. (2000). “Genetic heterogeneity in Noonan syndrome: Evidence for an autosomal recessive form”. American Journal of Medical Genetics. 94 (1): 46–51.
- 10. Schubbert S, Zenker M, Rowe SL, et al. (2006). “Germline KRAS mutations cause Noonan syndrome”. Nat. Genet. 38 (3): 331–6.
- 11. Bentires-Alj M, Kontaridis MI, Neel BG (2006). “Stops along the RAS pathway in human genetic disease”. Nat. Med. 12 (3): 283–5.
- Roberts AE, Araki T, Swanson KD, et al. (2007). “Germline gain-of-function mutations in SOS1 cause Noonan syndrome”. Nat. Genet. 39 (1): 70–4.
- 13. Razzaque MA, Nishizawa T, Komoike Y, et al. (2007). “Germline gain-of-function mutations in RAF1 cause Noonan syndrome”. Nat. Genet. 39 (8): 1013–7.
- 14. De Luca, A.; Bottillo, I.; Sarkozy, A.; Carta, C.; Neri, C.; Bellacchio, E.; Schirinzi, A.; Conti, E.; Zampino, G.; Battaglia, A.; Majore, S.; Rinaldi, M. M.; Carella, M.; Marino, B.; Pizzuti, A.; Digilio, M. C.; Tartaglia, M.; Dallapiccola, B. (2005).
- Noonan, JA (1968). “Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease”. Am. J. Dis. Child. 116 (4): 373–80.
سندرم بال پروانهای Epidermolysis Bullosa (EB) Syndrome
https://medlineplus.gov/genetics/condition/noonan-syndrome/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام