
سندرم مارشال
Marshall Syndrome


شاهین اسعدی (دانشجوی ژنتیک مولکولی)، مهسا جمالی (کارشناس ارشد ژنتیک)، رعنا باقری (کارشناس ارشد ژنتیک)، حمیده محمدزاده (کارشناس ارشد ژنتیک)، محیا فتاحی (کارشناس ارشد ژنتیک)
نگارنده مسئول: شاهین اسعدی (Molecular Geneticist)
کلیات
یک اختلال ژنتیکی بافت همبند است که میتواند منجر به از دست رفتن شنوایی در انسان گردد. سه منطقه شایع که تحت تأثیر سندرم مارشال قرار میگیرند عبارتند از چشمها، مفاصل و ساختار صورت


شکل 1: تصویر کودک مبتلا با ویژگیهای مشخص صورت
علائم و نشانههای بالینی
بیماران مبتلا به سندرم مارشال، دارای اختلالاتی در ساختار صورت هستند که شامل پل پهن بینی، فاصله گسترده چشمها از یکدیگر (هایپرتلوریسم)، چشمهای برجسته یا بزرگتر از حد نرمال و آب مروارید در چشمها میباشد. علاوه بر این، قسمت فوقانی جمجمه ضخیمتر از حد طبیعی است و کلسیم بیش از حد در جمجمه ذخیره میشود. از دست دادن حس شنوایی نیز در مبتلایان سندرم مارشال ممکن است رخ دهد که ناشی از حساسیت عصبی میباشد. علائم دیگری که در مبتلایان سندرم میتواند رخ دهد عبارتند از: چشمهای متقاطع (ازوتروپی)، هایپرتروپی یا وضعیتی که خط بینایی چشم در یک چشم بالاتر از چشم دیگر است، جدا شدن شبکیه چشم، افزایش فشار داخل چشم (گلوکوم)، پوسیدگی دندانهای فک بالا و استخوان بینی کوچکتر از حد طبیعی.
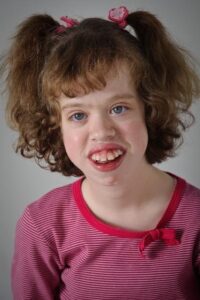
شکل 2: تصویر دختر مبتلا به سندرم مارشال همراه با ویژگیهای متمایز صورت و ناهنجاریهای دندان
علتشناسی
سندرم در اثر جهش ژن COL11A1 که در بازوی کوتاه کروموزوم شماره 1 بهصورت 1p21.1 مستقر است، ایجاد میشود. این ژن دستورالعمل لازم برای سنتز یک مؤلفه از پروتئین کلاژن نوع 11 را فراهم میکند که بهعنوان زنجیره پرو-آلفا 1 نیز شناخته میشود. کلاژنها مولکولهایی هستند که ساختار و استحکام بافتهای متصل به عضلات، مفاصل، اندام و پوست بدن را پوشش میدهند. کلاژن نوع 11 بهطور معمول در غضروف که در طول توسعه رشد، بیشترین قسمت استخوان را تشکیل میدهد، یافت میشود؛ بنابراین جهش در ژن COL11A1 منجر به تولید پروتئین ناقص یا غیرکاربردی از کلاژن نوع 11 میشود که در نتیجه آن، استحکام بافتها در بدن سست میشود و میتواند علائم و نشانههای سندرم مارشال را به وجود آورد.

شکل 3: نمای شماتیکی از کروموزوم شماره 1 که ژن COL11A1 در بازوی کوتاه این کروموزوم بهصورت 1p21.1 مستقر است
سندرم مارشال از الگوی توارثی اتوزومال غالب پیروی میکند؛ بنابراین برای ایجاد سندرم مارشال یک نسخه از ژن جهشیافته COL11A1 (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به سندرم مارشال در این حالت، برای هر بارداری احتمالی به میزان 50% میباشد. شایان ذکر است که سندرم مارشال در برخی موارد از الگوی توارثی اتوزومال مغلوب نیز تبعیت میکند؛ بنابراین برای ایجاد سندرم مارشال در حالت مغلوب اتوزومی، دو نسخه از ژن جهشیافته COL11A1 (یکی از پدر و دیگر از مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به سندرم مارشال در این حالت، برای هر بارداری احتمالی به میزان 25% است.
فراوانی
سندرم مردان و زنان را به تعداد مساوی تحت تأثیر قرار میدهد. فرکانس شیوع این سندرم تاکنون در هیچ پایگاه علمی گزارش نشده است.

شکل 4: تصاویری از کودک مبتلا به سندرم مارشال با ظاهری متمایز
تشخیص
سندرم بر اساس یافتههای بالینی مبتلایان و برخی آزمایشهای پاتولوژیکی، تشخیص داده میشود. دقیقترین روش تشخیص سندرم مارشال بهویژه از سایر سندرمها مانند سندرم استیکلر، آزمایش ژنتیک مولکولی برای ژن COL11A1 بهمنظور بررسی وجود جهشهای احتمالی میباشد.
مسیرهای درمانی
استراتژی درمان و مدیریت سندرم بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص پوست، متخصص چشم، متخصص گوش و حلق و بینی، متخصص ارتوپدی، جراح چشم، جراح پلاستیک و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان قاطعی برای این سندرم وجود ندارد و تمامی اقدامات پزشکی بهمنظور کاهش رنج مبتلایان میباشد. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند از اهمیت بسزایی برخوردار است.
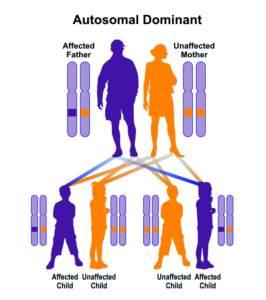
شکل 5: نمای شماتیک از الگوی توارثی اتوزومال غالب (سمت راست) و اتوزومال مغلوب (سمت چپ) که سندرم مارشال نیز از این الگوها پیروی میکند
منابع:
اسعدی شاهین، داداشپور مهدی، علیپور شهریار، جمالی مهسا، پورجعفر رؤیا، وحدانیکیا ویدا، فیروزی اکرم، باقری رعنا، سادهدل سمانه، قلیزاده زهرا، کتاب پاتولوژی در ژنتیک پزشکی (4) جلد چهارم (M-W)، صفحات 644-639، انتشارات کتب دانشگاهی عمیدی، زمستان 1396 (2018)
https://rarediseases.org/rare-diseases/marshall-smith-syndrome/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام