
سندرم اسلای
Sly syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، مهسا جمالی (کارشناس ارشد ژنتیک)، حمیده محمدزاده (کارشناس ارشد ژنتیک)

نگارنده مسئول: شاهین اسعدی (Molecular Geneticist)
کلیاتی از سندرم اسلای
سندرم اسلای یک اختلال ژنتیکی است که بهعنوان موکوپلیساکاریدوز نوع 7 (MPS VII) نیز شناخته میشود. موکوپلیساکاریدوز نوع 7، اختلال ژنتیکی و متابولیکی نادری است که بسیاری از نقاط بدن را تحت تأثیر میگذارد. ویژگیهای بالینی افراد مبتلا به موکوپلیساکاریدوز نوع 7 از فردی به فرد دیگر متفاوت است، اما همه آنها در کوتاهی قد و قامت به دلیل عقبماندگی رشد، ناهنجاریهای اسکلتی، تغییرات غیرطبیعی در استخوانها با پراش اشعه ایکس (X-Ray) و برخی از درجات ناتوانی فکری، مشترک هستند. بقاء در بزرگسالی برای مبتلایان به موارد خفیف از موکوپلیساکاریدوز نوع 7 رایج است و آرتروز از عوارض شایع نوع خفیف MPS VII میباشد.

شکل 1: تصویر کودک مبتلا به سندرم اسلای (MPS VII)
موکوپلیساکاریدوز نوع 7 نیز مانند سایر اختلالات موکوپلیساکاریدوز، در گروه بیماریهای ژنتیکی و متابولیکی ارثی بوده که بهعنوان اختلالات ذخیرهسازی لیزوزومی شناخته میشود. عملکرد لیزوزوم بهعنوان واحد گوارش اولیه در درون سلول است و آنزیمها در درون لیزوزوم شکسته شده و یا باعث هضم مواد مغذی خاص، مانند کربوهیدراتها و چربیها میشوند. در افراد مبتلا به اختلالات موکوپلیساکاریدوز از جمله موکوپلیساکاریدوز نوع 7، کمبود و یا عملکرد نادرست آنزیمهای لیزوزومی منجر به تجمع غیرطبیعی از کربوهیدراتهای پیچیده خاص در سلول (موکوپلیساکاریدها یا گلیکوز آمینوگلیکانها) در بافتهای مختلف مانند، اسکلت اندامها، مفاصل، مغز، ستون فقرات، بند ناف، قلب، طحال، کبد و غیره میشود. سندرم اسلای یا موکوپلیساکاریدوز نوع 7 (MPS VII)، بیماری ذخیره لیزوزومی است که با کمبود آنزیم بتا-گلوکورونیداز همراه است.

شکل 2: نمای شماتیک از ساختار حلقوی آنزیم بتا-گلوکورونیداز
علائم و نشانههای سندرم اسلای
موارد بسیار شدید از موکوپلی ساکاریدوز نوع 7، توسط هیدروپس فتالیس و یا وقتیکه مایع اضافی در بدن تجمع مییابد، قبل از تولد تشخیص داده میشود. این امر ممکن است باعث مرگ جنین در رحم مادر و یا مرگ بعد از تولد گردد و همچنین ممکن است در نوزاد مبتلا به MPS VII زردی پوست شبیه یرقان رخ دهد. کودکان با موارد خفیفتر از موکوپلیساکاریدوز نوع 7، علائم بیماری را در دوران کودکی نشان میدهند. کودکان مبتلا به MPS VII، ناتوانی در رشد تنه و کوتاه بودن قامت (کوتولگی تنه) را نشان میدهند. همچنین اندازه سر این کودکان ممکن است بیشازحد بزرگ باشد (ماکروسفالی) و گردن ممکن است بیشازحد کوتاه باشد.
شایان ذکر است انواع ناهنجاریهای متعدد استخوان (دیستوزیس) که در اغلب افراد مبتلا به موکوپلیساکاریدوز مشاهده میشود، در کودکان مبتلا به موکوپلیساکاریدوز نوع 7 (MPS VII) نیز رایج است. این تغییرات استخوانی ممکن است شامل یک استخوان برجسته پستان، دنده گشاد، دررفتگی مکرر مفصل ران، انقباض مفاصل (یخزدگی مفاصل یا بیحسی)، پاچنبری و زاویهدار شدن زانو به داخل (واروس زانو) یا زانوی پرانتزی باشد. همچنین ناهنجاری نخاعی نیز ممکن است وجود داشته باشد؛ از جمله انحنای ستون فقرات (اسکولیوز) و یا کیفوز که در برخی از مبتلایان سندرم اسلای دیده میشود. علاوه بر این، کودکان مبتلا به این اختلال ممکن است یک ظاهر غیرمعمول با صورت درشت را داشته باشند و در سنین 7 ماهگی تا 8 سالگی ممکن است کدورت ابری در قرنیه چشم نیز رخ دهد. برخی از مبتلایان به این اختلال ممکن است تأخیر رشد در زبان و گفتار و ناتوانی فکری را آشکار کنند، این در حالی است که اکثر مبتلایان سندرم اسلای دارای هوش طبیعی هستند.
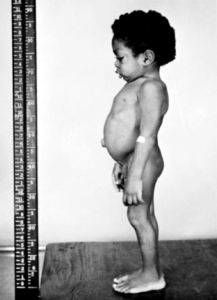
شکل 3: تصویر کودک مبتلا به سندرم اسلای، به کوتاهی قامت و شکم حجیم و بیرونزدگی ناف از شکم دقت کنید
در موکوپلیساکاریدوز نوع 7 (MPS VII)، ممکن است کبد و یا طحال بهطور غیرطبیعی بزرگ شود و فتق شکمی یا برآمدگی شکم رخ دهد. در برخی از کودکان مبتلا به این اختلال، رودهها ممکن است از طریق دیواره شکم در منطقه ناف (فتق ناف) بیرونزدگی داشته باشند. بعلاوه برخی از کودکان مبتلا به سندرم اسلای، ممکن است از دست دادن عمیق حس شنوایی، عفونتهای مکرر گوش میانی و دستگاه تنفسی فوقانی، ضخیم شدن بافت نرم گلو و یا تارهای صوتی، بزرگ شدن غیرطبیعی زبان (بزرگی زبان)، مشکلات قلبی (مانند سوفل قلبی و نارسایی آئورت) و پرمویی بیشازحد بدن (هایرسوتیسم) را تجربه کنند.
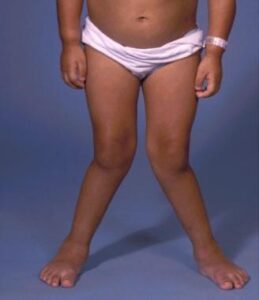
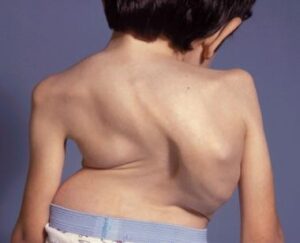
شکل 4: تصاویر کودکان مبتلا به سندرم اسلای همراه با حالت پرانتزی زانوها و کیفوزاسکولیوز یا ناهنجاری غیرطبیعی ستون فقرات
متوسط امید به زندگی در مبتلایان نوع خفیف سندرم اسلای، بین 19 تا 20 سال تخمین زده میشود. میزان امید به زندگی در این بیماران، به دلیل عفونتهای مکرر دستگاه تنفسی فوقانی، عوارض عصبی و اختلالات دستگاه گوارشی کاهش مییابد. لازم به ذکر است که یک فرم خفیف از موکوپلیساکاریدوز نوع 7، در ادبیات پزشکی گزارش شده است که علائم بیماری در طول دهه دوم زندگی فرد بیمار آغاز شده است. در این بیماران، علائم اختلال به نظر میرسد بسیار کمتر از حد معمول MPS VII کلاسیک است و ممکن است شامل تغییرات استخوانی جزئی و درشتی بسیار خفیف صورت باشد. بزرگی طحال و کبد در این فرم از موکوپلیساکاریدوز نوع 7 (MPS VII) گزارش نشده است.
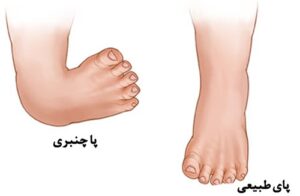
شکل 5: نمای شماتیک از پای طبیعی و پاچنبری که در مبتلایان سندرم اسلای مشهود است
علتشناسی سندرم اسلای
سندرم اسلای یا همان موکوپلیساکاریدوز نوع 7 (MPS VII)، در اثر جهش ژن GUSB که در بازوی بلند کروموزوم شماره 7 بهصورت 7q11.21 مستقر است، ایجاد میشود. این جهش منجر به کمبود یا نقص در سنتز آنزیم لیزوزومی به نام بتا-گلوکورونیداز میشود. جهش ژن GUSB ممکن است در ایجاد علائم و نشانههای بالینی مبتلایان سندرم اسلای از فردی به فرد دیگر متفاوت باشد. آنزیم بتا-گلوکورونیداز در تجزیه مولکول قند درشت به نام گلیکوزآمینوگلیکانها (GAG) ایفای نقش میکند، بنابراین کمبود آنزیم بتا-گلوکورونیداز منجر به تجمع این مولکولها در سلولها از طریق لیزوزوم میشود. تجمع گلیکوزآمینوگلیکانها در سلولها منجر به بزرگ شدن یا افزایش حجم بافتها و اندامها میگردد.
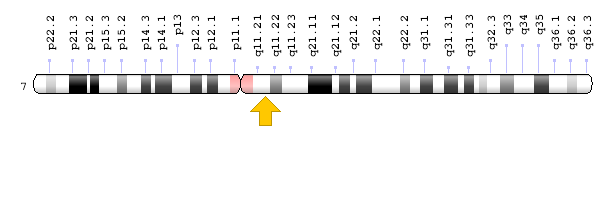
شکل 6: نمای شماتیک از کروموزوم شماره 7 که ژن GUSB در بازوی بلند این کروموزوم بهصورت 7q11.21 مستقر است
سندرم اسلای یا موکوپلیساکاریدوز نوع 7 (MPS VII) از الگوی توارثی اتوزومال مغلوب پیروی میکند. همانطور که میدانید در اختلالات ژنتیکی مغلوب، زمانی فرد بیمار میشود که دو کپی یا دو نسخه از ژن جهشیافته را یکی از پدر و یکی از مادر به ارث ببرد، پس برای ایجاد سندرم اسلای نیز دو نسخه از ژن جهشیافته GUSB موردنیاز است.
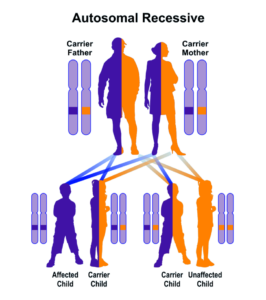
شکل 7: نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم اسلای نیز از این الگو پیروی میکند
فراوانی سندرم اسلای
سندرم اسلای یا همان موکوپلیساکاریدوز نوع 7 (MPS VII) بسیار نادر است. فرکانس شیوع این اختلال حدود 1 در 250000 تولد زنده تخمین زده میشود. تابهحال کمتر از 100 مورد از سندرم اسلای در ایالات متحده آمریکا گزارش گردیده است. سندرم اسلای زنان و مردان را به تعداد مساوی تحت تأثیر قرار میدهد.
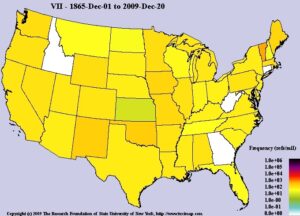
شکل 8: نمای شماتیک از فرکانس شیوع سندرم اسلای (MPS VII) در جهان
تشخیص سندرم اسلای
اولین اقدام برای تشخیص اختلال موکوپلیساکاریدوزها، آزمایش ادرار است که میزان درماتانسولفات، هپارانسولفات و کندروئیتینسولفات که در ادرار افراد مبتلا به اختلالات موکوپلیساکاریدوز این ترکیبات افزایش مییابد را مورد ارزیابی قرار میدهند. تشخیص موکوپلیساکاریدوز نوع 7 (MPS VII) توسط یک ارزیابی بالینی کامل و دقیق که شامل شرححال دقیق بیمار و آزمون تخصصی برای اندازهگیری سطح آنزیم بتا-گلوکورونیداز در سلولهای خونی و پوستی میباشد، انجام میپذیرد. آزمایش ژنتیک مولکولی برای بررسی جهش در ژن GUSB بهمنظور تأئید تشخیص سندرم اسلای، قطعیترین تست میباشد، همچنین تشخیص پیش از تولد نیز از طریق آمنیوسنتز و یا نمونهبرداری از پرزهای جفتی جنین برای اندازهگیری فعالیت آنزیم بتا-گلوکورونیداز و یا آزمایش ژنتیک مولکولی برای بررسی جهش در ژن GUSB جهت تأئید تشخیص، امروزه امکانپذیر است.
مسیرهای درمانی سندرم اسلای
مسیرهای درمانی موکوپلیساکاریدوز نوع 7 (MPS VII) علامتی و حمایتی است. تغییرات استخوانی و فتق نیز ممکن است به جراحی نیاز داشته باشد. اختلالات چشمی (کدورت قرنیه) و قلبی عروقی نیز ممکن است با عمل جراحی درمان شوند. بیماران مبتلا به اختلالات موکوپلیساکاریدوز ممکن است به دلیل ناهنجاری در راه هوایی دستگاه تنفسی فوقانی و یا ستون فقرات گردن، به بیهوشی حساسیت داشته باشند، بنابراین احتیاطهای لازم بایستی قبل از عمل جراحی به این بیماران اعمال شود. مشاوره ژنتیک برای خانوادههای دارای فرزند یا سابقه خانوادگی مبتلا به موکوپلیساکاریدوز نوع 7 (MPS VII) نیز توصیه میشود. درمان آنزیمی جایگزین نیز امروزه امکانپذیر است. اولین مورد درمان آنزیمی جایگزین در سال 2014 با تجویز آنزیم rhGUS به دوزاژ 2mg/kg بهصورت هفتهای یکبار، تحت درمان قرار گرفت. این بیمار پس از 24 هفته تجویز منظم آنزیم جایگزین، بدون عوارض جانبی درمان شد. در طی فرایند درمان با این آنزیم، سطح تجمع گلیکوزآمینوگلیکانها و موکوپلیساکاریدها در ادرار کاهش یافت و اندازه طحال و کبد به میزان 60% کاهش یافت و عملکرد ریوی (دستگاه تنفسی) بهبود یافت. درمان آنزیمی جایگزین هنوز هم در ایالات متحده آمریکا ادامه دارد.

شکل 9: تصویر داروی سلکترازیم جهت درمان جایگزینی آنزیم بتا-گلوکورونیداز
تاریخچه سندرم اسلای
اختلال موکوپلیساکاریدوز نوع 7 (MPS VII) اولین بار توسط دکتر ویلیام شافورد اسلای پزشک متخصص بیوشیمی و زیستشناسی مولکولی William Shuford Sly (19. Oktober 1932 in East St. Louis) در سال 1973 گزارش گردید.

شکل 10: تصویر دکتر ویلیام شافورد اسلای، کاشف سندرم اسلای در سال 1973
منابع:
اسعدی شاهین، جمالی مهسا، باقری رعنا، سادهدل سمانه، توحیدی راد مانوش، کتاب پاتولوژی در ژنتیک پزشکی جلد دوم (M-Z)، صفحات 1132-1125، انتشارات کتب دانشگاهی عمیدی، بهار 1396
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام