
آپرت سندرم
Apert syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، دکتر مسعود ملکی (سیتولوژیست)، دکتر علی نظیرزاده (متخصص ژنتیک مولکولی)، الناز حیدری (دانشجوی کارشناسی ارشد ژنتیک)
نگارنده مسئول: شاهین اسعدی (Geneticist)

سندرم آپرت، یک اختلال ژنتیکی مادرزادی با ناهنجاری جمجمه، صورت، دستها و پاها (آکروسفالوسینداکتیلی) میباشد. این بیماری را بهعنوان سندرم قوس برونشی (حلق) نیز طبقهبندی میکنند. سندرم آپرت، اولین بار در سال 1906 توسط پزشک فرانسوی به نام دکتر یوجین چالرز آپرت گزارش گردید، البته در گذشتههای باستان نیز این بیماری وجود داشت و نام آن آکروسفالوسینداکتیلی بود. آکرو در زبان یونان باستان به معنای اوج و سفالو به معنای سر و سینداکتیلی به معنای بافتهشدن انگشتان دست و پا به همدیگر است. در علم جنینشناسی، بافتهای همبند سلولهای دست و پا بهصورت انتخابی جهت فرگشت صحیح دست و پا و داشتن فرم طبیعی، دچار آپوپتوزیس یا همان مرگ فیزیولوزیک سلولی میشوند، اما در جنینهایی که استعداد سندرم آپرت را دارند، آپوپتوزیس در سلولهای مزبور رخ نمیدهد و به همین دلیل بافتهای انگشتان دست و پا به همدیگر چسبیده میمانند.


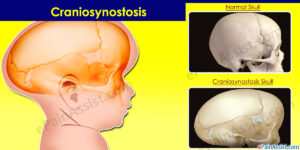
افراد مبتلا به سندرم آپرت، دچار کرانیوسینوستوزیس (قوز شدن جمجمه و استخوانهای صورت) میشوند. البته با اعمال جراحی در جمجمه و استفاده از نخهای ترومای (بخیه) ویژه، میتوان الگوی رشد مناسبی برای فرم طبیعی جمجمه فراهم کرد.
فرکانس سندرم آپرت در بین جمعیتهای مختلف، متفاوت است و تخمین زده میشود که شیوع این بیماری 1 در 160000 تولد یا 1 در 200000 تولد باشد.
علائم و نشانههای بالینی سندرم آپرت
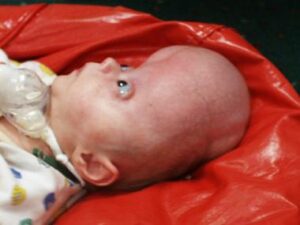
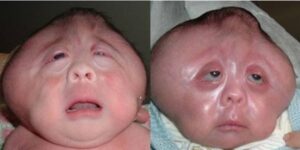
ناهنجاری جمجمه آشکارترین اثرات آکروسفالوسینداکتیلی را نشان میدهد. کرانیوسینوستوزیس در سندرم آپرت قطعاً رخ میدهد که در آن جمجمه خیلی سریعتر از رشد ارگانیک کودک، فرگشت مییابد، هرچند که مغز کودک هنوز در حال رشد و گسترش است. پیشانی برجسته و صاف بودن پشت جمجمه از دیگر نشانههای سندرم آپرت است، همچنین کمبود میزان هوشیاری به عبارتی عقبماندگی ذهنی، رشد ناقص استخوانهای صورت، چشمان برآمده از حدقه، دهان باز و زبان از دهان بیرون زده، گوشهای برجسته به جلو، فرگشت ناقص دندانها و بدریختی دندانها و بهم چسبیدن انگشتان دست و پا از نشانههای آشکار سندرم آپرت نیز میباشند.
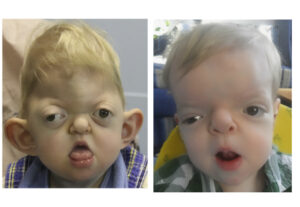
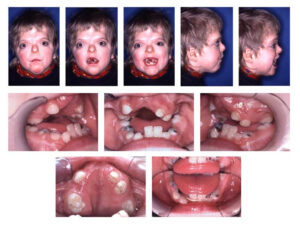
همه مبتلایان سندرم آپرت در آکروسفالوسینداکتیلی باهم مشترک هستند؛ اما ناهنجاریهای فرم دستها در مبتلایان سندرم آپرت، همیشه نشاندهنده چهار ویژگی مشترک است:
(a) انگشت شست کوتاه با انحراف شعاعی
(b) بهمچسبیدگی انگشت اشاره و فرم حلقه مانند داشتن
(c) چسبندگی میان تمام انگشتان دست و پا
(d) چسبندگی ساده در فضای چهارم دستها
بدشکلی از فضای بین انگشت شست تا انگشت اشاره ممکن است متغیر باشد.
ژنتیک مولکولی سندرم آپرت
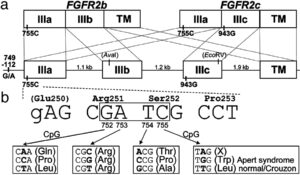
سندرم آپرت، یک اختلال ژنتیکی مادرزادی است که از الگوی توارثی اتوزومال غالب پیروی میکند. حدود 70% از موارد سندرم آپرت، به دلیل جهش در ژن FGFR2 است که منجر به جابجایی نوکلئوتید سیتوزین بجای گوانین در موقعیت 755 سنتز پروتئین فیبروبلاست میشود. این فرآیند، یک کانون جهش خاص در مردان است؛ به عبارتی مطالعات در این زمینه نشان میدهد که این جهش ژنتیکی اغلب در منشأ آلل پدری رخ میدهد. بروز شدت بیماری آپرت، با افزایش سن پدر نیز افزایش مییابد. مطالعات در آنالیز اسپرم، نشان میدهد که مردان مسن با استعداد سندرم آپرت دارای جهش ژن FGFR2 در محتوای اسپرم خود هستند.
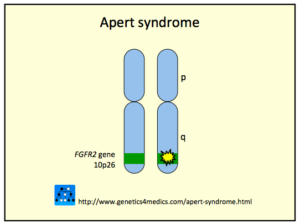
ژن FGFR2 در بازوی بلند کروموزوم شماره 10 بهصورت 10q26 مستقر است و مسئول سرکوب، فرآیند آپوپتوزیس و نگهدارنده مزانشیم بین انگشتی است.
مسیرهای درمانی سندرم آپرت
جراحی برای فرم طبیعی جمجمه و رشد کامل مغز بهصورت ارگانیک، امری ضروری است. جراحی دندانها نیز برای تکامل صحیح و طبیعی موردنیاز است. در حال حاضر هیچ درمان استانداردی برای چسبندگی انگشتان دست و پا در سندرم آپرت وجود ندارد. هر بیمار بایستی بسته بهشدت بیماریش روشها و قابلیتهای استفاده بهینه از دست و پای خودش را فرابگیرد. تشویق خانواده و دوستان برای بهبود عملکرد دستها و پاهای این بیماران بسیار حائز اهمیت است چراکه انسان بیمار خیلی سریع ناامید میشود و این حالت یعنی مرگ تدریجی برای بیماران خاص، اما حمایت و حس همدردی و همچنین عدم بیتوجهی به چنین بیمارانی میتواند بسیار امیدوارکننده برای آنها باشد.
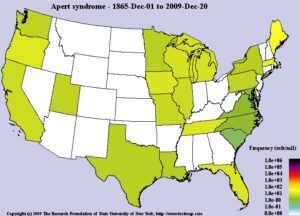
نقشه پراکندگی بیماران مبتلا به سندرم آپرت در جهان از سال 1865 تا سال 2009

References:
- Fearon, Jeffrey A. (July 2003). “Treatment of the Hands and Feet in Apert Syndrome: An Evolution in Management”. Plastic and Reconstructive surgery (Dallas, Texas) 112 (1): 1–12.
- Abe Bert Baker; Lowell H. Baker (1979). “Apert’s Syndrome”. Clinical Neurology (Medical Dept., Harper & Row) 3: 47.
- Kaplan, L C (April 1991). “Clinical assessment and multispecialty management of Apert syndrome”. Clinics in plastic surgery 18 (2): 217–25.
- Upton, J (April 1991). “Apert Syndrome. Classification and pathologic anatomy of limb anomalies”. Clinics in plastic surgery 18 (2): 321–55.
- Herman, TE; Siegel, MJ (October 2010). “Apert syndrome with omphalocele”. J Perinatol. 30 (10): 695–697.
- Ercoli, G; Bidondo, MP; Senra, BC; Groisman, B (September 2014). “Apert syndrome with omphalocele: a case report”. Birth Defects Res A Clin Mol Teratol. 100 (9): 726–729.
- Wilkie, A O; S F Slaney; M Oldridge; M D Poole; G J Ashworth; A D Hockley; R D Hayward; D J David; L J Pulleyn; P Rutland (February 1995). “Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome”. Nature Genetics 9 (2): 165–72.
- Goriely, A.; McVean, GA; Röjmyr, M; Ingemarsson, B; Wilkie, AO (2003). “Evidence for Selective Advantage of Pathogenic FGFR2 Mutations in the Male Germ Line”. Science 301 (5633): 643–6.
- Moloney, DM; Slaney, SF; Oldridge, M; Wall, SA; Sahlin, P; Stenman, G; Wilkie, AO (1996). “Exclusive paternal origin of new mutations in Apert syndrome”. Nature Genetics 13 (1): 48–53.
- Britto, J A; J C T Chan; R D Evans; R D Hayward; B M Jones (May 2001). “Differential expression of fibroblast growth factor receptors in human digital development suggests common pathogenesis in complex acrosyndactyly and craniosynostosis”. Plastic and Reconstructive surgery 107 (6): 1331–1338.
- Hajihosseini MK, Duarte R, Pegrum J, Donjacour A, Lana-Elola E, Rice DP, Sharpe J, Dickson C (February 2009). “Evidence that Fgf10 contributes to the skeletal and visceral defects of an Apert syndrome mouse model”. Dev. Dyn. 238 (2): 376–85.
- Zucker, R M (April 1991). “Syndactyly correction of the hand in Apert syndrome”. Clinics in plastic surgery 18 (2): 357–64.
سندرم آلستروم
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام