
سندرم میکروفتالمی آهینیا بوسما
Bosma Arhinia Microphthalmia Syndrome
(B.A.M Syndrome)

شاهین اسعدی ( دانشجوی ژنتیک مولکولی)
کلیاتی از سندرم B.A.M
سندرم B.A.M یک اختلال ژنتیکی نادر است که با اختلالات چشم، بینی و مشکلات بلوغ جنسی همراه است. ویژگیهای اصلی سندرم B.A.M معماری چهره است که با عدم وجود بینی (دماغ) خارجی مشخص میشود. در حالی که اکثر افراد مبتلا به سندرم B.A.M بدون بینی متولد میشوند، برخی از این مبتلایان نیز ممکن است بینی بهشدت کم توسعهیافته را (هایپوپلاستیک) داشته باشند. علاوه بر این، افراد آسیبدیده با این سندرم ممکن است حس بویایی نداشته باشند و بنابراین افراد مبتلا به سندرم B.A.M بوی هیچ چیزی را حس نخواهند کرد.
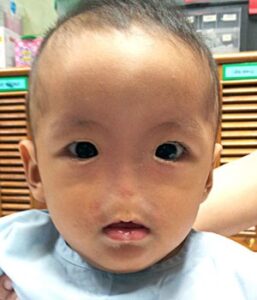
شکل 1: تصویر کودک مبتلا به سندرم B.A.M همراه با اختلالات چشمی و عدم وجود خارجی بینی
علائم و نشانههای بالینی سندرم B.A.M
در اکثر افراد مبتلا به سندرم B.A.M، چشمها یا بهطور غیرطبیعی کوچک (میکروفتالمی) هستند و یا اصلاً وجود ندارند (آنوفتالمی) که باعث اختلال بینایی یا نابینایی میشود. اختلالات اضافی چشم در سندرم B.A.M شامل وجود شکاف یا سوراخ در ساختار چشم (کلوبوم) و اَبری شدن لنزهای چشم (آب مروارید) میباشند.

شکل 2: تصاویری از کودکان مبتلا به سندرم B.A.M همراه با ویژگیهای مشخص صورت
اختلالات اضافی سر و صورت که ممکن است در افراد مبتلا به سندرم B.A.M رخ دهد عبارتند از: شکاف کام، عدم وجود سینوسها در پشت بینی (سینوسهای پارانازال)، انسداد سوراخهای بینی (آترزی کوآنال)، تنگی مجاری اَشک و کوچکی فک بالای دهان.
بسیاری از این ناهنجاریها بهویژه در نوزادان آسیبدیده منجر بهدشواری تنفس میشود. برخی از افراد مبتلا به سندرم B.A.M دارای گوش خارجی غیرطبیعی هستند.
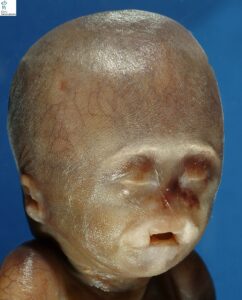
شکل 3: تصویر نوزاد مبتلا به سندرم B.A.M همراه با اختلالات چشمی (عدم وجود بافت چشم باز) و عدم وجود خارجی بینی و لب شکافی و اختلالات گوشها و گردن
افراد مبتلا به سندرم B.A.M همچنین دارای هایپوگنادیسم هایپوگنادوتروپیک هستند که یک اختلال ناشی از کاهش تولید هورمونهای جنسی است. بدون درمان، این اختلالات هورمونی اغلب باعث تأخیر در بلوغ جنسی میشود. مردان مبتلا به سندرم B.A.M نیز ممکن است دارای بافتهای جنسی غیرتوسعهیافته و عدم فرود بیضهها به داخل کیسه بیضه باشند.
علت شناسی سندرم B.A.M
سندرم B.A.M معمولاً در اثر جهش ژن SMCHD1 که در بازوی کوتاه کروموزوم شماره 18 بهصورت 18p11.32 مستقر است، ایجاد میشود. این ژن دستورالعملهای لازم برای سنتز پروتئینی را فراهم میکند که در تنظیم بیان ژن با تغییر ساختار DNA ایفای نقش میکند. بهطور خاص، پروتئین SMCHD1 نقش مهمی در خاموش کردن برخی از ژنها دارد. در میان دیگر توابع، پروتئین SMCHD1 به نظر میرسد برای توسعه بینی، چشم و دیگر ساختارهای سر و صورت مهم باشد. هنوز مشخص نیست که جهش در ژن SMCHD1 چگونه بر عملکرد پروتئین SMCHD1 تأثیر میگذارد و منجر به اختلال در توسعه ساختارهای سر و صورت مربوط به سندرم B.A.M میشود. تصور میشود که تغییرات در این ژن منجر به خاموش شدن غیرطبیعی ژنهای درگیر در توسعه سر و صورت میشود که میتواند منجر به میکروفتالمی و سایر اختلالات مشخصشده در سندرم B.A.M شود. مشکلات ناشی از توسعه بینی ممکن است نورونهای هورمون آزادکننده گنادوتروپین (GnRH) را تحت تأثیر قرار دهد که سلولهای عصبی هستند و آزاد شدن هورمونهای تولیدمثل را کنترل میکنند. نورونهای GnRH در بینی در حال رشد هستند و سپس به مغز منتقل میشوند. اختلال رشد این نورونها میتواند هایپوگنادیسم هایپوگنادوتروپیک را در افراد مبتلا به سندرم B.A.M ایجاد کند.
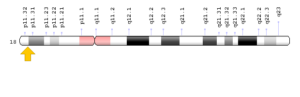
شکل 4: نمای شماتیک از کروموزوم شماره 18 که ژن SMCHD1 در بازوی کوتاه این کروموزوم بهصورت 18p11.32 مستقر است
سندرم B.A.M در بیشتر موارد به دلیل جهشهای جدید و بدون سابقه خانوادگی ایجاد میشود؛ اما در برخی موارد نیز سندرم B.A.M از الگوی توارثی اتوزومال غالب پیروی میکند؛ بنابراین برای ایجاد این سندرم به یک نسخه از ژن جهشیافته SMCHD1 (اعم از پدر یا مادر) نیاز است و شانس داشتن فرزندی مبتلا به سندرم B.A.M در حالت اتوزومی غالب، برای هر بارداری احتمالی به میزان 50% میباشد.
فراوانی سندرم B.A.M
سندرم B.A.M اختلال ژنتیکی بسیار نادر است که فرکانس شیوع آن در جهان مشخص نیست. تاکنون کمتر از 100 مورد مبتلا به سندرم B.A.M از سراسر جهان در ادبیات پزشکی گزارش شده است.
تشخیص سندرم B.A.M
سندرم B.A.M بر اساس یافتههای بالینی و فیزیکی مبتلایان و برخی آزمایشهای پاتولوژیکی، تشخیص داده میشود. دقیقترین روش تشخیص سندرم B.A.M، آزمایش ژنتیک مولکولی برای ژن SMCHD1 بهمنظور بررسی وجود جهشهای احتمالی میباشد. تشخیص پیش از تولد نیز با استفاده از تکنیک PGD و مایع آمنیوسنتز و یا نمونهبرداری از پرزهای کوریونی جفت جنین امکانپذیر است.
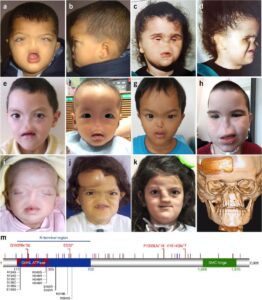
شکل 5: تصاویری از مبتلایان سندرم B.A.M همراه با اختلالات مربوطه
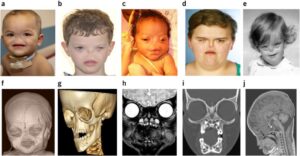
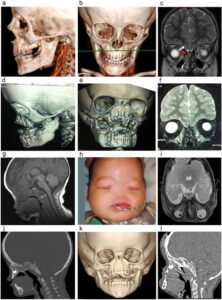
شکل 6: تصاویر رادیولوژیکی از اختلالات مربوطه در مبتلایان سندرم B.A.M
مسیرهای درمانی سندرم B.A.M
استراتژی درمان و مدیریت سندرم B.A.M بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص چشم، متخصص فک و دهان، متخصص جراحی زیبایی، متخصص و جراح گوش و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان استانداردی برای این سندرم وجود ندارد و تمام اقدامات بالینی بهمنظور تخفیف رنج مبتلایان میباشد. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند، ضرورت دارد.
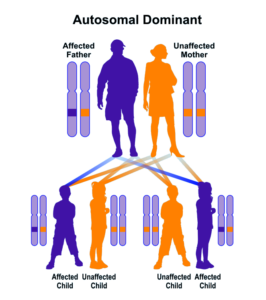
شکل 7: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم B.A.M نیز از این الگو تبعیت میکند
منبع:
اسعدی شاهین، ملتیار حسن، روشنروان ندا، جمالی مهسا، دادشپور مهدی، محمدزاده حمیده، فتاحی محیا، کتاب پاتولوژی در ژنتیک پزشکی 7 (A-H)، فصل 2، صفحات 132-123، انتشارات کتب دانشگاهی عمیدی، بهار 1397
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام