
سندرم آندرسون فابری
Anderson-Fabry Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، دکتر علی نظیرزاده (متخصص ژنتیک مولکولی)، الناز حیدری (دانشجوی کارشناسی ارشد ژنتیک)


نگارنده مسئول: شاهین اسعدی (Geneticist)
سندرم فابری، بهعنوان بیماری فابری، بیماری اندرسون فابری، اختلال آنژیوکراتوماکورپوریس و یا بیماری کمبود آلفاگالاکتوزیداز شناخته میشود. سندرم اندرسون فابری، یک بیماری نادر ژنتیکی است که در اثر اختلال ذخیره لیزوزومی ایجاد میشود. سندرم اندرسون فابری از الگوی توارثی وابسته به X مغلوب تبعیت میکند. بیماری فابری میتواند، طیف گستردهای از علائم سیستمیک را شامل شود که پیامد آن اختلال در ترکیب شیمیایی اسفنگولیپیدها و در پی آن سوختوساز ناکارآمد ترکیبات بیوشیمیایی بدن خواهد بود. سندرم اندرسون فابری، اولین بار توسط دو متخصص پوست به نامهای یوهانس فابری از آلمان و ویلیام اندرسون از انگلستان در سال 1898 گزارش گردید.


سمت راست، دکتر ویلیام اندرسون و سمت چپ، دکتر یوهانس فابری
علائم و نشانههای بالینی سندرم فابری اندرسون
علائم این بیماری معمولاً در اوایل دوران کودکی بروز میکند، اما درک این بیماری و تشخیص آن در زمان کودکی دشوار خواهد بود. این علائم شامل:
حس درد: بیماران مبتلا به سندرم فابری اندرسون، معمولاً تجربه درد موضعی یا کامل اندامهای بدن را دارند و یا اختلال در دستگاه گوارش در این بیماران رایج میباشد. اعتقاد بر این است که این حس درد در اندامها، به آسیب رشتههای عصبی محیطی که انتقال پیام حس درد را برعهده دارند، مرتبط است. حس درد در دستگاه گوارش، به احتمال زیاد توسط تجمع چربی در عروق کوچک لنفاوی ایجاد میشود که منجر به مسدود شدن راه جریان خون میگردد.
اختلال در کلیهها: عوارض کلیوی شامل نقص عملکرد کلیه و نارسایی کلیه میباشد که در اکثر بیماران مبتلا به سندرم فابری اندرسون مشترک است، این عوارض جدی و تهدیدکننده حیات بیماران میباشد و ممکن است در طول زندگی شدت این علائم بیشتر شود. وجود بیش از حد پروتئین در ادرار که باعث ادرار کفآلود میشود، اولین نشانه از درگیری کلیهها در سندرم فابری اندرسون است. بیشترین اختلالات کلیوی در این بیماران در دهه سوم عمر آنها ایجاد میشود که یک علت شایع مرگ ناشی از این بیماری است.
تظاهرات بالینی قلبی عروقی در سندرم فابری اندرسون
عوارض قلبی در اثر اختلال، در گلیکولیپیدها ایجاد میشود که با افزایش سن، میزان خطر بیماریهای قلبی و عروق کرونری بیشتر میشود. در این بیماران، فشارخون بالا و کاردیومیوپاتی معمولاً مشاهده میگردد.

تظاهرات پوستی در سندرم فابری اندرسون
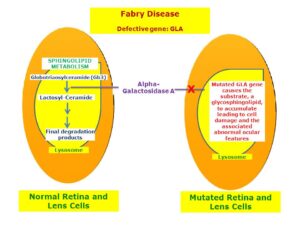
این تظاهرات شامل آنژیوکراتوماس یا پاپولهای کوچک و بدون درد است که میتواند در هر منطقه از بدن ظاهر شود، اما اغلب بر روی ران، اطراف ناف، باسن، شکم و کشاله ران شایعتر هستند. آنهیدروزیس یا عدم تعریق بدن، یک نشانه شایع جهت مشکوک بودن به بیماری فابری اندرسون است، علاوه بر این بیماران مبتلا به سندرم فابری اندرسون علائمی مانند بیماری رینود با نوروپاتی را نشان میدهند که بهطور خاص منجر به حس درد و سوزش اندامها میشود. از علائم چشمی در این بیماری میتوان به کدورت قرنیه، ورم ملتحمه، اختلالات عروقی شبکیه بهصورت قدامی/خلفی مانند آب مروارید نیز اشاره کرد.
سایر تظاهرات بالینی در سندرم فابری اندرسون
خستگی، نوروپاتی، درد همراه با سوزش در اندامها، اختلال عروق مغزی که منجر به افزایش خطر سکته مغزی میشود، وزوز گوش، سرگیجه، حالت تهوع، ناتوانی در افزایش وزن، عدم تعادل شیمیایی و اسهال از دیگر علائم سندرم فابری اندرسون میباشند.
نمای شماتیک از اختلال وزوز کردن گوش انسان در بیماران مبتلا به سندرم فابری اندرسون

نمای شماتیک دینامیکی از حالت دیدگان همراه با سرگیجه در بیماران مبتلا به سندرم فابری اندرسون
پاتوفیزیولوژی سندرم فابری اندرسون
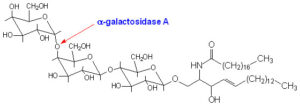
کمبود آنزیم آلفاگالاکتوزیداز A با توجه به جهش در ژن GLA باعث تجمع گلیکولیپیدها در عروق خونی شده که این تجمع منجر به اختلال در عملکرد مناسب بافتهای بدن میشود. جهش در ژن GLA برای ایجاد بیماری فابری اندرسون از الگوی توارثی وابسته به X مغلوب پیروی میکند که با نفوذ ناکامل این ژن، در زنان هتروزیگوت همراه است، بنابراین زنان حامل این جهش، از نظر ژنتیک جمعیت نسبت به بیماری فابری اندرسون، هتروزیگوس خواهند بود. مردان دارای جهش در ژن GLA که این ژن تغییریافته را از مادر ناقلشان دریافت کردهاند، نسبت به بیماری فابری اندرسون از نظر ژنتیک جمعیت، هموزیگوس بوده و بیماری فابری اندرسون را بروز خواهند داد.
شایان ذکر است که زنان حامل ژن جهشیافته GLA، نیز در مواردی مستعد بیماری فابری اندرسون هستند اما درجه و شدت علائم این بیماری در زنان خفیفتر از مردان است. تحقیقات جدید نشان میدهد که بسیاری از زنان دچار سندرم فابری اندرسون از مشکلاتی چون آب مروارید چشم در اوایل بیماری مربوطه، احتمال سکته مغزی، اختلالات هایپرتروفیک بطن چپ قلب و نارسایی کلیوی نیز رنج میبرند.
ژنتیک مولکولی سندرم فابری اندرسون
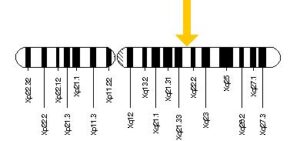
همانطور که ذکر شد، سندرم فابری اندرسون توسط جهش در ژن GLA ایجاد میشود. ژن GLA در بازوی بلند کروموزوم جنسی X بهصورت Xq22.1 مستقر است. جهش در این ژن باعث جابجایی نوکلئوتید گوانین بهجای سیتوزین شده که همین امر منجر به ترجمه نادرست اسید آمینه آسپارژین به اسید آمینه سرین در موقعیت 215 برای سنتز پروتئین میگردد.
این جهش ژنتیکی با الگوی توارثی وابسته به X مغلوب منتقل میشود. این جهش باعث کمبود یا عدم وجود آنزیم آلفاگالاکتوزیداز شده که همین امر منجر به بروز اختلالات ذخیرهای لیزوزومی میگردد.

تشخیص سندرم فابری اندرسون
بیماری فابری اندرسون، براساس تظاهرات بالینی فرد مظنون تشخیص داده میشود. بدین ترتیب که با مشاهده علائم بالینی فرد مشکوک به سندرم فابری اندرسون، میتوان با اندازهگیری آنزیم آلفاگالاکتوزیداز بر روی لکوسیتها برای تشخیص صحیح بیماری فابری اندرسون استفاده کرد.
در زنان مشکوک به سندرم فابری اندرسون برای تشخیص بیماری مذکور، با توجه به ماهیت غیرفعال بودن یکی از کروموزومهای جنسی X در زنان، استفاده از روش ارزیابی آنزیمی قابلاعتماد نخواهد بود. تجزیهوتحلیل ژنتیک مولکولی ژن GLA، از دقیقترین روشهای تشخیص برای زنان مشکوک به سندرم فابری اندرسون است. بیوپسی کلیه نیز، اگر تجمع بیش از حد چربی را نشان دهد ممکن است مطرحکننده بیماری فابری اندرسون باشد. متخصصین اطفال و داخلی معمولاً بیماری فابری اندرسون را نمیتوانند با علائم بالینی ذکرشده، تشخیص دهند، بنابراین تشخیص قطعی و کاملاً صحیح برعهده متخصص ژنتیک مولکولی است.
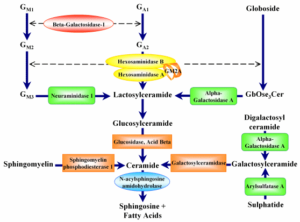 مسیر درمانی سندرم فابری اندرسون
مسیر درمانی سندرم فابری اندرسون
اولین راه درمانی برای سندرم فابری اندرسون، توسط سازمان غذا و داروی آمریکا FDA در 24 آوریل سال 2003 به تصویب رسید. داروی فابرازیم (بتاآگالزیداز) یا آلفاگالاکتوزیداز از طرف شرکت داروسازی Genzyme مجوز تولید گرفت. این دارو، یک مسیر درمانی برای جایگزین کردن آنزیم از دست رفته در اثر سوء عملکرد ژنتیکی بیماران فابری اندرسون است، اما هزینه خرید این دارو بسیار گران است، بطوریکه هزینه استفاده سالانه داروی فابرازیم در سال 2012 در ایالات متحده آمریکا به ازای هر بیمار، 200000 دلار بود که غیرقابل استفاده برای بسیاری از بیماران در سرتاسر جهان بدون بیمه و شرایط اقتصادی مطلوب میباشد.
داروی فابرازیم که بهمنظور جایگزین کردن آنزیم آلفاگالاکتوزیداز مورد استفاده قرار میگیرد، درمان قطعی نیست ولی میتواند سوختوساز بدن را بهبود بخشد و تا حدودی از پیشرفت بیماری جلوگیری کند، اما درمان قطعی سندرم فابری اندرسون محسوب نمیشود. شایان ذکر است که استفاده از روش جایگزین آنزیمی (ERT) به کمک داروی فابرازیم ممکن است همراه با دردهای عضلانی عصبی و یا دردهای التهابی همراه باشد، بنابراین مصرفکنندههای این دارو میبایست از داروهای ضددرد و ضدتشنج و ضدالتهاب نیز استفاده کنند.
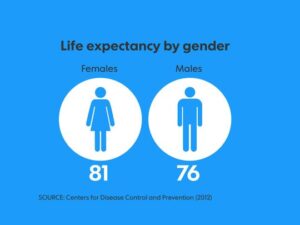
پیشبینی متوسط زندگی در سندرم فابری اندرسون
متوسط امید به زندگی با بیماری فابری اندرسون در مردان مبتلا، 58/2 سال میباشد، درحالیکه متوسط امید به زندگی در جمعیت عمومی مردان، 74/7 سال است و متوسط امید به زندگی در زنان مبتلا به سندرم فابری اندرسون، 75/4 سال است درحالیکه متوسط امید به زندگی در جمعیت عمومی زنان، 80/0 سال میباشد. با استناد به اطلاعات بیوانفورماتیک سازمان بهداشت جهانی، از سال 2001 تا سال 2008، شایعترین علت مرگومیر در مبتلایان سندرم فابری اندرسون، در ارتباط با بیماریهای قلبی عروقی و پیوند کلیه بوده است.
اپیدمیولوژی سندرم فابری اندرسون
فرکانس بروز سندرم فابری اندرسون در جهان حدود 1 تولد در هر 40000 تولد و 1 تولد در هر 120000 تولد زنده انسان است. این متغیر بودن فرکانس سندرم فابری اندرسون، به اکوسیستم افراد و رژیم غذایی و سبک زندگی افراد مستعد به بیماری فابری اندرسون بستگی دارد.

References:
- Hoffmann، Bjoern; Beck، Michael; Sunder-Plassmann، Gere; Borsini، Walter; Ricci، Roberta; Mehta، Atul (2007). “Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy—a retrospective analysis from the Fabry Outcome Survey”.
- Chew، E.; Ghosh، M.; McCulloch، C. (June 1982). “Amiodarone-induced cornea verticillata”. Canadian Journal of Ophthalmology 17 (3): 96–99.
- Marchesoni، Cintia L.; Roa، Norma; Pardal، Ana María; Neumann، Pablo; Cáceres، Guillermo; Martínez، Pablo; Kisinovsky، Isaac; Bianchi، Silvia; Tarabuso، Ana Lía; Reisin، Ricardo C. (May 2010). “Misdiagnosis in Fabry disease”.
- Fervenza، Fernando C.; Torra، Roser; Warnock، David G. (December 2008) [13 November 2008].
- Keating، Gillian M. (October 2012). “Agalsidase alfa: a review of its use in the management of Fabry disease”. BioDrugs 26 (5): 335–354.
- Waldek، Stephen; Patel، Manesh R.; Banikazemi، Maryam; Lemay، Roberta; Lee، Philip (November 2009). “Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry”. Genetics in Medicine 11 (11): 790–796.
- Mehta، A.; Ricci، R.; Widmer، U.; Dehout، F.; Garcia de Lorenzo، A.; Kampmann، C.; Linhart، A.; Sunder-Plassmann، G.; Ries، M.; Beck، M. (March 2004). “Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey”. European Journal of Clinical Investigation 34 (3): 236–42.
- James، William D.; Berger، Timothy G.; Elston، Dirk (2006). Andrews’ Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN0-7216-2921-0.
- Schiffmann، Raphael; Kopp، Jeffrey B.; Austin، Howard A.; Sabnis، Sharda; Moore، David F.; Weibel، Thais; Balow، James E.; Brady، Roscoe O. (June 2001). “Enzyme replacement therapy in Fabry disease: a randomized controlled trial”. JAMA 285 (21): 2743–2749.
- Wilcox، William R.; Banikazemi، Maryam; Guffon، Nathalie; Waldek، Stephen; Lee، Philip; Linthorst، Gabor E.; Desnick، Robert J.; Germain، Dominique P. (July 2004).
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید