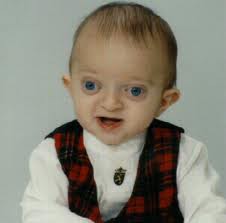
سندرم آلاژیل
Alagille Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، دکتر روشنک سامبرانی (متخصص ژنتیک)، دلشاد عبداللهنیا (کارشناس ارشد بیوتکنولوژی)، مهسا جمالی (دانشجوی کارشناسی ارشد ژنتیک)

نگارنده مسئول: شاهین اسعدی (Geneticist)
سندرم آلاژیل یک اختلال ژنتیکی است که کبد، قلب، کلیه و سیستمهای دیگر بدن را تحتتأثیر قرار میدهد. مشکلات در ارتباط با این اختلال در اوایل دوران کودکی آشکار میشود. سندرم آلاژیل از الگوی وراثتی اتوزومال غالب پیروی میکند و فرکانس این بیماری 1 در هر 100000 تولد زنده میباشد. این بیماری برای اولین بار در سال 1970 توسط دکتر دنیل آلاژیل متخصص کبد و دستگاه گوارشی گزارش گردید.


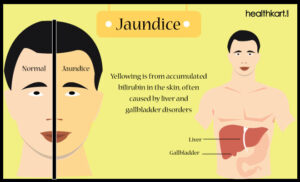
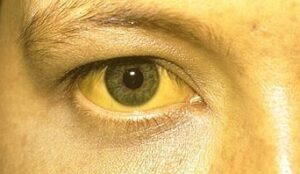
شدت این اختلال از خفیف تا شدید، مثلاً در پیوند قلب یا کبد بسته به سن فیزیولوژیک اهداء کننده نیز متغیر است. علائم و نشانههای ناشی از آسیب کبدی در سندرم آلاژیل ممکن است شامل یک اثر خفیف از رنگ زرد در پوست و سفیدی چشم (یرقان)، خارش و تجمع کلسترول در پوست باشد.
بیوپسی کبد در سندرم آلاژیل ممکن است مجاری صفروای را در چند نقطه مسدود و یا در برخی موارد انسداد کامل مجاری صفراوی را نشان دهد. علاوه بر هیپوپلازی (تشکیل نشدن کامل) مجاری صفراوی داخل کبدی که منجر به کلستاز میشود، نوزادان مبتلا به این نشانگان (سندرم)، ممکن است دچار آنومالیهای صورت، قلب (بصورت تنگی دریچه ریوی یا تنگی محیطی عروق ریوی و تترالوژی فالوت)، ستون فقرات (مهره پروانهای)، چشم (امبریوتوکسین خلفی) و کلیه (دیسپلازی کلیه) باشند. بیماران در نتیجه آن دچار سطح کلسترول خونی بالا و گزانتومهای پوستی همراه آن هستند. رشد ناکافی و عقبماندگی ذهنی خفیف هم گاهی وجود دارد.
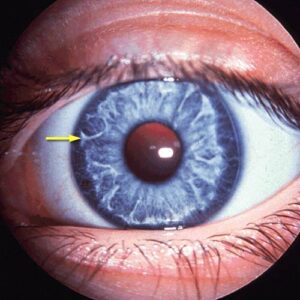

تترالوژی فالوت یک نقص قلبی عروقی شایع در مبتلایان سندرم آلاژیل است. این نقص از چهار اختلال جداگانه تنگی جداره عروق ریوی ( دستگاه تنفسی ) یا انسداد عروق ریوی، اختلال آئورت، نقص دیواره بین بطنی و هایپرتروفی بطن راست تشکیل شده است. محدوده مرگومیر در صورت عدم درمان تترالوژی فالوت در مبتلایان با درجه سنی 10 سال به میزان %70 و در افراد بالای 40 سال به میزان %95 میباشد، با این حال عمل جراحی کامل و دقیق میتواند بهطور قابلتوجهی طول عمر و کیفیت زندگی بیماران مبتلا به سندرم آلاژیل را بهبود بخشد. افراد مبتلا به سندرم آلاژیل دارای ویژگیهای مشترک در صورت از جمله پیشانی برجسته، چشمهای درشت و چانه کوچک هستند.
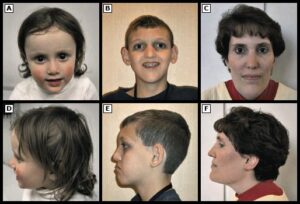
ژنتیک مولکولی سندرم آلاژیل
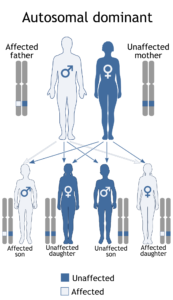
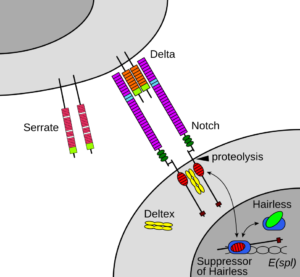
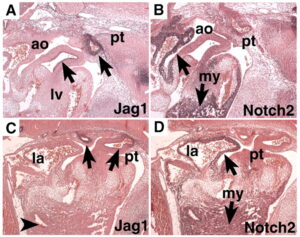
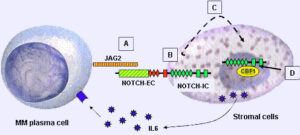
سندرم آلاژیل در اثر جهش ژنهای JAG1 و NOTCH2 ایجاد میشود. این اختلال از الگوی اتوزومال غالب تبعیت میکند؛ بدین معنا که فقط یک کپی از ژنهای جهشیافته مذکور برای ایجاد سندرم آلاژیل کافی است.
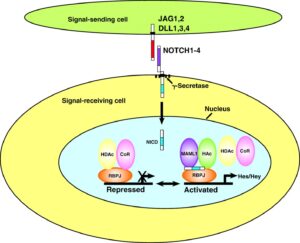
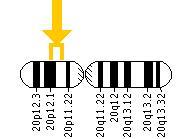
این سندرم حتی در میان خانوادههایی که سابقه سندرم آلاژیل را ندارند نیز رخ میدهد. ژن JAG1 در بازوی کوتاه کروموزوم شماره 20 بصورت 20p12.1،20p12.2 مستقر است. این ژن مسئول تنظیم سرنوشت سلول در بسیاری از سیستمهای بدن است که عامل فعال شدن مسیر پروتئولیتیک در سلولهای بدن میباشد.
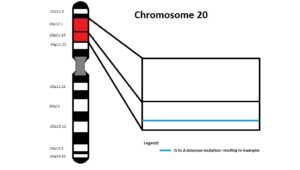
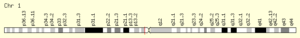
ژن NOTCH2 در بازوی کوتاه کروموزوم شماره 1 بصورت 1p13.3 مستقر است. این ژن در سیستم عصبی مسئول مسیر سیگنالینگ سلول در تبادل اطلاعات دستگاه گلژی جهت تنظیم فرایندهای بیولوژیکی میباشد، همچنین این ژن ممکن است در عروق کرونری، کلیهها و کبد نیز ایفای نقش داشته باشد.
مسیر درمانی سندرم آلاژیل
در حال حاضر هیچ درمان شناختهشدهای برای سندرم آلاژیل وجود ندارد، اما از درمانهای در دسترس میتوان به بهبود عملکرد قلب، کاهش اثرات اختلالات کبدی، کلیهها و بهبود عملکرد طحال نیز اشاره کرد.
برای بهبود عملکرد صفرا از داروی اورسودیول میتوان استفاده کرد. برای کاهش خارش پوست میتوان از هیدروکسیزین، کلوستیرامین، ریفامپیسین، فنوباربیتال و نالترکسون استفاده کرد. همچنین بسیاری از بیماران مبتلا به سندرم آلاژیل برای بهبود عملکرد صفرا از دوزهای بالای مولتیویتامین که شامل ویتامینهای A،D،E،K است نیز استفاده میکنند.
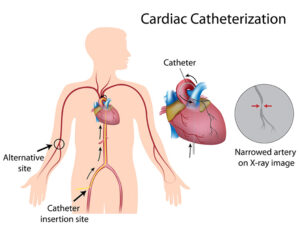
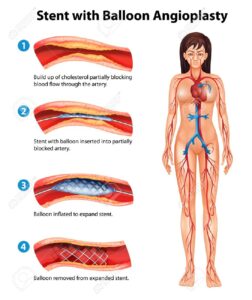
جراحی نیز گاهی اوقات برای ترمیم نقص قلبی در بیماران مبتلا به سندرم آلاژیل مورداستفاده قرار میگیرد. همچنین به دلیل شریانهای ریوی باریک در مبتلایان سندرم آلاژیل، اغلب یک فرآیند کاتتریزاسیون مشابه آنژیوپلاستی برای گسترش عروق کرونری بهمنظور کاهش فشار در سمت راست قلب ممکن است مورداستفاده قرار گیرد. در موارد متوسط تا شدید سندرم آلاژیل، ممکن است در شریانها برای افزایش قطرشان، استنتگذاری شود. پیوند کبد در موارد شدید سندرم آلاژیل جایگزین مناسبتری نسبت به داروها است.
References:
- Alagille D، Odièvre M، Gautier M، Dommergues JP (January 1975). “Hepatic ductular hypoplasia associated with characteristic facies، vertebral malformations، retarded physical، mental، and sexual development، and cardiac murmur”. J. Pediatr. 86 (1): 63–71.
- Oda، T.، Elkahloun، A.G.، Pike، B.L.، Okajima، K.، Krantz، I.D.، Genin، A.، Piccoli، D.A.، Meltzer، P.S.، Spinner، N.B.، Collins، F.S. and Chandrasekharappa، S.C.، 1997. Mutations in the human Jagged1 gene are responsible for Alagille syndrome.، Nat. Genet.، pp. 235-242.
- Kamath، B.M.، Bauer، R.C.، Loomes، K.M.، Chao، G.، Gerfen، J.، Hutchinson، A.، Hardikar، W.، Hirschfield، G.، Jara، P.، Krantz، I.D.، Lapunzina، P.، Leonard، L.، Ling، S.، Ng، V.L.، Hoang، P.L.، Piccoli، D.A. and Spinner، N.B.، 2012. NOTCH2 mutations in Alagille syndrome. J Med Genet 49، 138-44.
- Rosalie David A، Contis G (1996). “Paleopathology on schistosomiasis in Egyptian mummies”. Parasitol. Today (Regul. Ed.) 12 (4): 167.
- H S.J. Lee، ed. (1999). Dates in Gastroenterology: A Chronological Record of Progress in Gastroenterology over the Last Millennium (Landmarks in Medicine). Informa Healthcare.
- Moodley J; Singh B; Lalloo S; Pershad S; et al. (2001). “Non-operative management of haemobilia”. The British journal of surgery 88 (8): 1073–6.
- van de Laar FA، Bor H، van de Lisdonk EH. (2008). “Prevalence of zebras in general practice: data from the Continuous Morbidity Registration Nijmegen.”. Eur J Gen Pract. 14. Suppl 1 (s1): 44–6.
- Adams، M.; Smith، U. M.; Logan، C. V.; Johnson، C. A. (2008). “Recent advances in the molecular pathology، cell biology and genetics of ciliopathies”. Journal of Medical Genetics 45 (5): 257–267.
- Davenport، J. R. (2005). “An incredible decade for the primary cilium: A look at a once-forgotten organelle”. AJP: Renal Physiology 289 (6): F1159–F1169.
- Ballbriga، Angel (1991). “One century of pediatrics in Europe (section: development of pediatric hospitals in Europe)”. In Nichols، Burford L.; et al. History of Paediatrics 1850–1950. Nestlé Nutrition Workshop Series 22. New York: Raven Press. pp. 6–8.
https://medlabnews.ir/%d8%b3%d8%b1%d8%b7%d8%a7%d9%86-%da%a9%d8%a8%d8%af/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام