روشها و تازههای تشخیص سندرمهای تالاسمی و اختلالات هموگلوبین
قسمت اول
دکتر حبیباله گلافشان
محمد اسماعیل خدمتی
دانشگاه علوم پزشکی شیراز
روشهای رایج الکتروفورز:
الکتروفورز کاپیلاری:
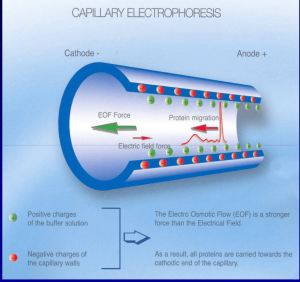
در این روش، الکتروفورز در لولههای کاپیلاری با قطر ۲۵ تا ۱۰۰ میکرومتر پر شده از بافر انجام میشود. جداسازی بر اساس اختلاف سرعت (Migration Velocity) یونهاست ولی مهمتر از آن حرکت مایع در کاپیلاری تحت اثر حرکت الکترواسموتیک است.
داخل لوله کاپیلاری، گروههای قابل یونیزه (Silanol group) است که به سرعت در دیواره ایجاد شارژ منفی میکنند. شارژ منفی دیواره موجب جذب شارژ مثبت یونهای بافر گردیده و ایجاد یک ابر دو لایه الکتریکی و اختلاف پتانسیل نزدیک جدار لوله میکند. زمانی که ولتاژ قوی در طول کاپیلاری برقرار شود، مولکولهای قسمت داخلی لوله (Diffuse layer) به طرف کاتد رفته و همراه خود مایع را با خود حرکت میدهند که نتیجه آن حرکت الکترواسموتیک و جدا شدن فراکسیونهای مختلف هموگلوبین است.
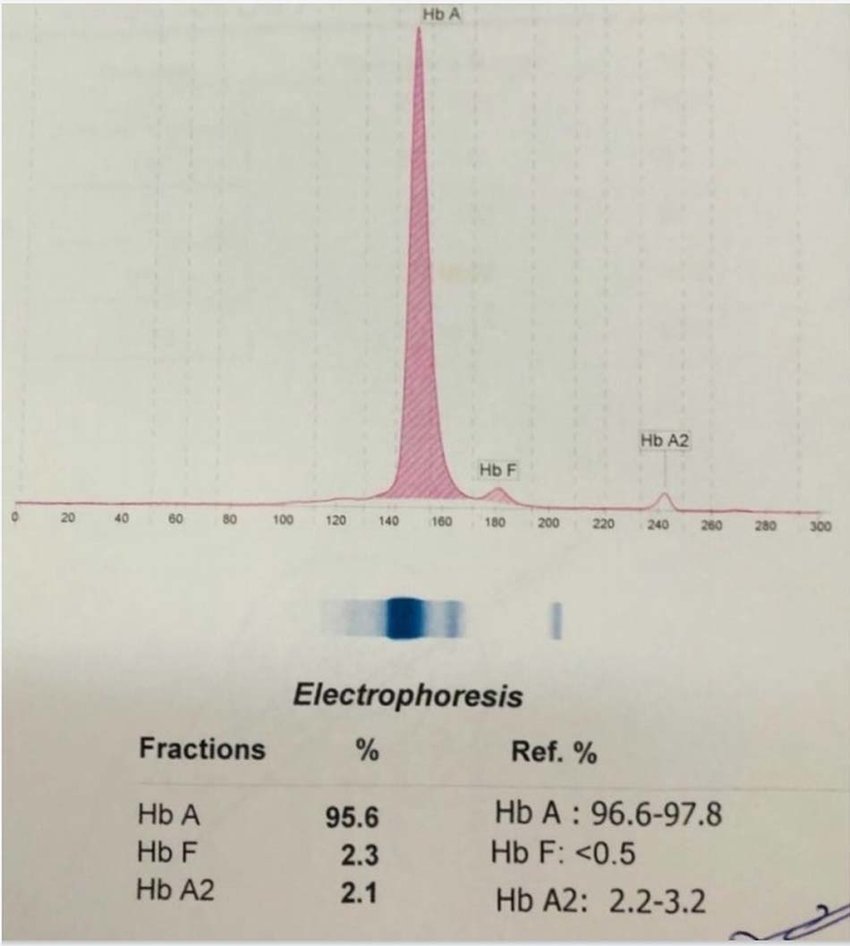
در الکتروفورز کاپیلاری فرایند الکتروفورز در بافر قلیایی با PH بالا در کاپیلاریهای سیلیکونی با قطر داخلی 50 میکرومتر انجام میگیرد؛ بدین مفهوم که نمونه به یک طرف کاپیلاری تزریق شده و تحت اثر حرکت الکترواسموتیک و ولتاژ قوی 10000 ولتی در طول کاپیلاری موجب جدا شدن فراکسیونهای مختلف هموگلوبین میشود.
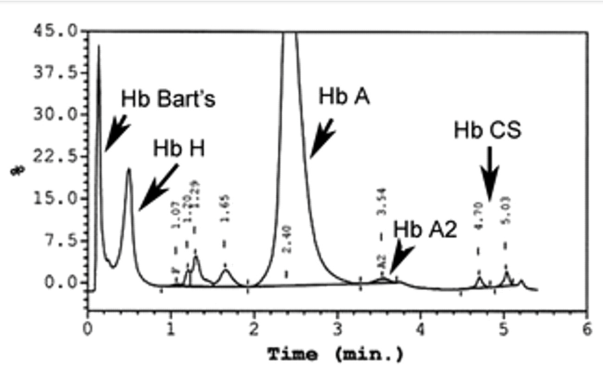
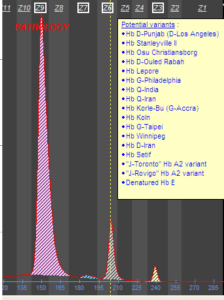
فراکسیونها با طول موج 415 نانومتر از کاتد به سوی آند به ترتیب هموگلوبینهای C، A2، E، S ،D، F، A، Bart، J، H قرائت میشود. برخلاف سیستم فشاری روش HPLC در این حالت حرکت الکترواسموتیک (EOF) در طول کاپیلاری پخش یکنواخت دارد و افت فشار در طول لوله مشاهده نمیشود. نمودار الکتروفورز بر روی محور مختصات رسم میگردد. محور Y غلظت و محور X بر اساس ناحیه حرکت (Migration Zone) کالیبره میگردد.
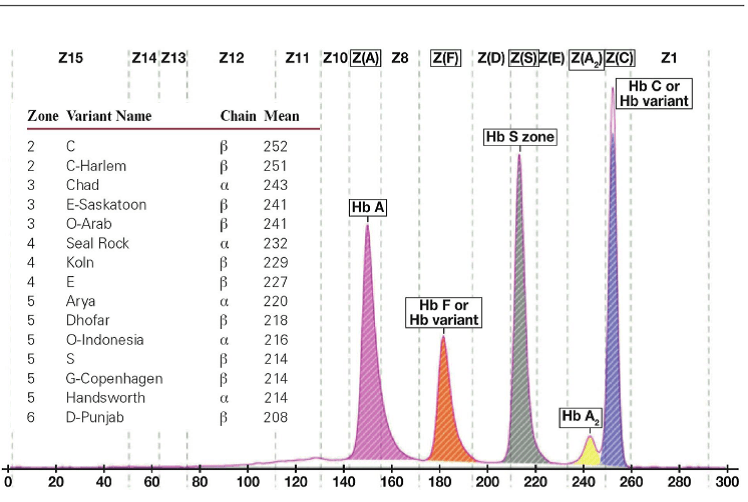
جداسازی هموگلوبینهای یک نمونه کنترل با هموگلوبینهای A و F و S و A2 و C با الکتروفورز موئینه مشاهده میشود. در الکتروفوروگرام محور X بر اساس عدد مهاجرت ناحیهبندی شده است و سطح زیر منحنی بر اساس ارتفاع پیک غلظت را نشان میدهد. عدد مهاجرت هموگلوبین A و A2 استاندارد بوده و حرکت هموگلوبینهای دیگر بر اساس حرکت هموگلوبین A مقایسه میگردد. واریانتهای هموگلوبین در 15 ناحیه مختلف قرار میگیرند.
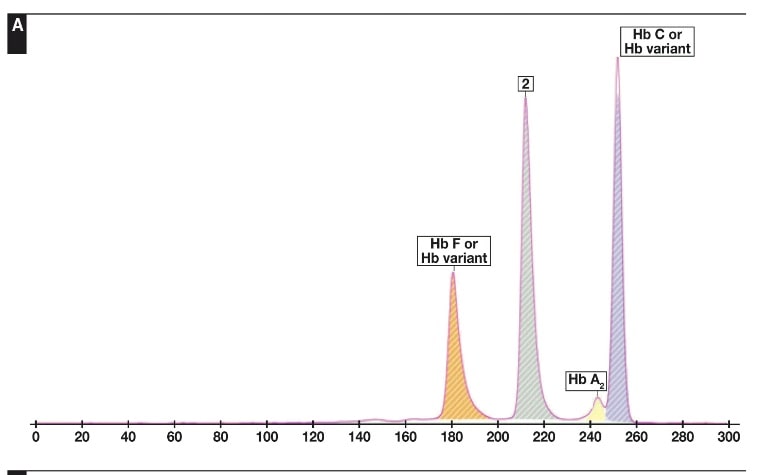
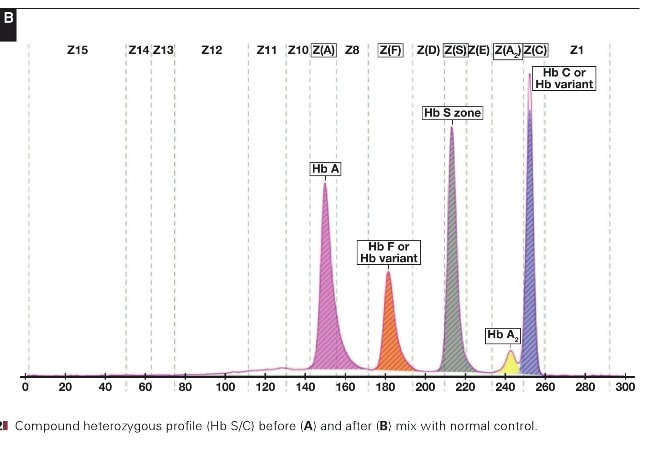
با توجه به این که جایگاه واریانهای هموگلوبین با توجه به عدد ناحیه هموگلوبین A مقایسه میشود، از این رو چنانچه الکتروفورز یک نمونه فاقد هموگلوبین A باشد، بایستی نمونه با خون نرمال مخلوط شود و الکتروفورز مجدد انجام شود تا عدد جایگاه A تأیید گردد.
برای مثال عدد مهاجرت هموگلوبین A برابر 150 و عدد جایگاه A2 عدد 250 و مربوط به هموگلوبین آریا 220 و مربوط به هموگلوبین S عدد 214 و … است. چنانچه در الکتروفورز یک نمونه باند A مشاهده نشود بایستی نمونه را با خون نرمال مخلوط کرد و باندهای غیرطبیعی نمونه را با توجه به مهاجرت باند A مورد تأیید قرار داد.
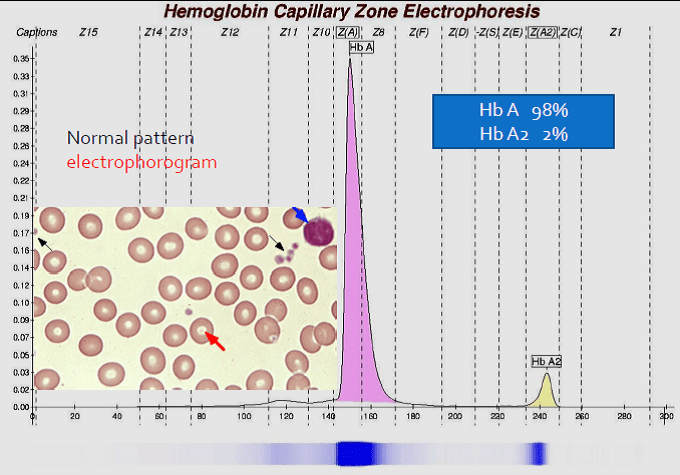

جایگاه قرار گرفتن واریانتهای مختلف هموگلوبین با الکتروفورز موئینه همراه با الکتروفورز یک نمونه نرمال
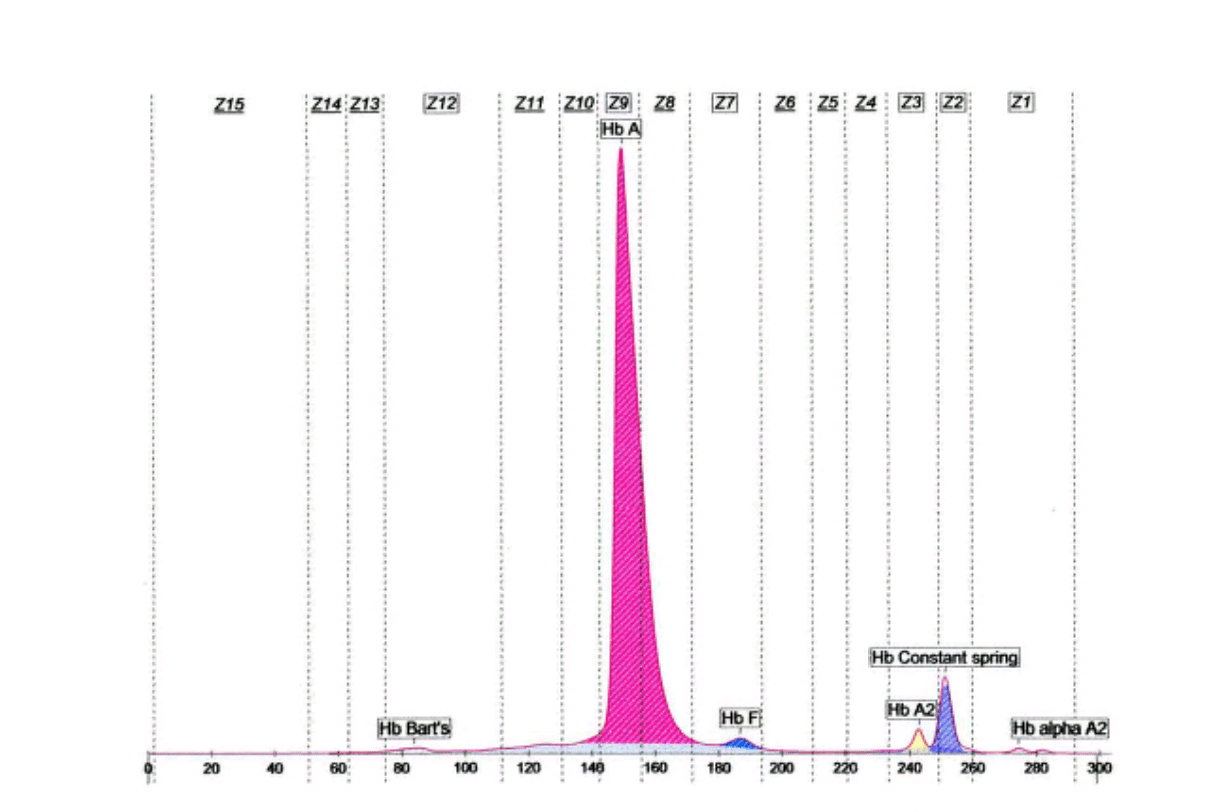
واریانتهای کنستانت اسپرینگ و A’2 به علت مقدار اندک، قابل تشخیص با روشهای الکتروفورز روی استات سلولز و ژل آگار نبوده و تشخیص آنها با این روشها نیاز به آپلیکاسیون غلیظ همولیزات دارد ولی روشهای کاپیلاری و کروماتوگرافی مایع با قابلیت بالا با سهولت بهتری این واریانها را آشکار میکند؛ برای مثال همراهی هموگلوبین کنستانت اسپرینگ در همراهی با حذف دو ژن آلفا ایجاد کمخونی بسیار شدید هموگلوبین اچ باکنستانت اسپیرینگ (H-CS) میکند که با روشهای کاپیلاری و کروماتوگرافی مایع با قابلیت بالا قابل تشخیص است. هموگلوبین A’2 که در محاسبه کل هموگلوبین A2 برای تشخیص تالاسمی ماینور لازم است با این دو روش قابل تشخیص است.
کروماتوگرافی مایع با قابلیت بالا (HPLC):
با هدف اندازهگیری هموگلوبینهای F و A2 و سایر واریانهای هموگلوبین ابداع گردیده است. این روش جداسازی دارای یک فاز جامد (Stationary phase) است که از ذرات رزین سه تا پنج میکرومتری شارژدار مثبت یا منفی پر شده است و یک فاز مایع (Mobile phase) در غلظتهای مختلف یونی است که تحت فشار از ستون جامد میگذرد.
در جداسازی انواع هموگلوبین با کروماتوگرافی، تبادل کاتیونی ترکیبات یک مخلوط پروتئینی جذب فاز شارژدارجامد ستون شده و با بهرهگیری از یک فاز متحرک مایع با فشار بالا ترکیبات نمونه را بر اساس درجه پیوند و رقابت با شارژهای ستون جدا میکند.
برای مثال در کروماتوگرافی تعویض کاتیونی (Cation exchane) ذرات فاز جامد دارای شارژ منفی هستند. نمونه خون به ستون تزریق شده و هموگلوبینها با اختلاف شارژی که دارند جذب ستون میشوند. شارژ مثبت قوی هموگلوبین موجب اتصال محکم آن به ستون و شارژ مثبت کمتر موجب اتصال ضعیف آن میشود. در مرحله بعد بافر با غلظت فزاینده کاتیونی تحت فشار به ستون هدایت میشود، شارژهای مثبت بافر برای اتصال به شارژهای منفی ستون رقابت کرده و از این رو موجب جدا شدن فراکسیونهای مختلف هموگلوبین بر اساس میل ترکیبی آن با ستون میگردند.
هموگلوبین اچ و بارت به طور سریع خارج و هموگلوبین C و S که شارژ مثبت بیشتری دارند کندتر خارج میگردند و مورد قرائت دستگاه قرار میگیرد. واریان های مختلف هموگلوبین در زمانهای مختلف در ارتباط با چگونگی پیوند با ستون، از ستون خارج میشوند. فاصله زمانی پیوند با ستون تا خارج شدن توسط بافر را RT یا Retention time گویند. کروماتوگرافی بهصورت پیکهایی در محور مختصات ظاهر میشود که محور x بر حسب RT و محور yبیانگر قله منحنی است و سطح زیر منحنی بیانگر غلظت هموگلوبین است.
در این روش الکتروفورز، فرمهای گلیکوزیله و غیر گلیکوزیله و نیز فرم هموگلوبین F استیله از غیر استیله دارای RT متفاوت بوده و جداگانه ایجاد پیک میکنند. واریانهای مختلفی نیز دارای RT یکسان هستند؛ برای مثال هموگلوبینهای A2 و لپور و E دارای RT یکسانند.
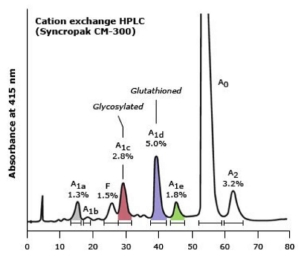
هموگلوبینهای قندی و مشتقات دناتوره دارای زمان خروج متفاوت بوده و جداگانه از ستون خارج میشوند
هموگلوبین قندی A1C به طور جداگانه ایجاد پیک میکند و میتوان غلظت آن را در غیاب واریانهای دیگر هموگلوبین به دست آورد و مقدار بیشتر از ۶ درصد دارای اهمیت بالینی است. بایستی توجه داشت که هموگلوبین S قندی در روش HPLC در جایگاه A قرار میگیرد و از این رو بیمار با کمخونی داسی که تزریق خون نداشته در محل A دارای باند کوتاه میگردد. در هتروزیگوت داسی مقدار هموگلوبین S حدود ۷ درصد کمتر گزارش میگردد، چون S قندی در جایگاه A قرار میگیرد.
بایستی توجه داشت که تزریق خون در هموگلوبینوپاتیها موجب ظاهر شدن باند A میگردد و از طرفی چون خون در دوران نگهداری با غلظت بالای گلوکز در تماس بوده است موجب افزایش هموگلوبین A1C میشود. بیلیروبین ایجاد باند در نواحی هموگلوبین H و بارت میکند، از این نمونه ژاندیس بایستی شسته و سپس به دستگاه داده شود. جدول زیر زمان RT و پیکهای مختلف هموگلوبین را با کروماتوگرافی HPLC نشان میدهد.
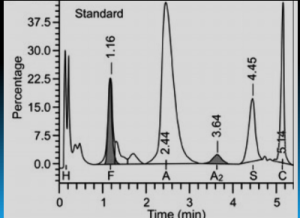
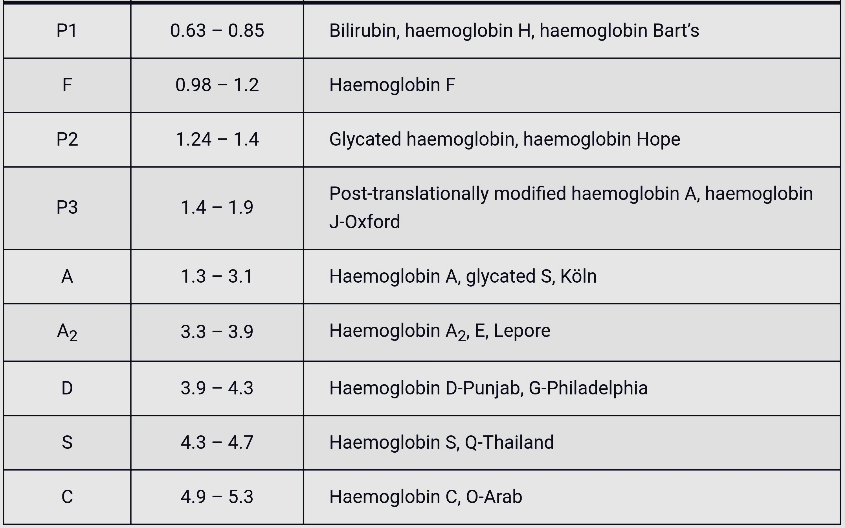
واریانهای مختلف هموگلوبین در زمانهای مختلف در ارتباط با چگونگی پیوند با ستون، از ستون خارج میشوند. فاصله زمانی پیوند با ستون تا خارج شدن توسط بافر را RT یا Retention time گویند. کروماتوگرافی بهصورت پیکهایی در محور مختصات ظاهر میشود که محور x بر حسب RT و محور y بیانگر قله منحنی است و سطح زیر منحنی بیانگر غلظت هموگلوبین است.
هموگلوبیـنهای S and C با شارژ مثبت بیشتری که دارند پیوند محکمتری با ستون داشته و زمان خروج آنها دیرتر است. هموگلوبینهای and bart H زود از ستون خارج شده و گرچه قابل شناسایی هستند ولی قابل اندازهگیری نیستند. در تابلوی بالا زمان خروج واریانتهای مختلف قید شده است.
الکتروفورز روی استات سلولز در PH قلیایی که به عنوان روش مادر از آن یاد میشود، یک روش قدیمی است که جداسازی هموگلوبینها روی استات سلولز در PH=8.2-8.6 صورت میگیرد. در این PH، هموگلوبینها دارای شارژ منفی میگردند و به طرف آند حرکت میکنند. برای هر ست الکتروفورز بایستی از کنترلهای A+F+S+C استفاده کرد که برای این کار میتوان خون نوزاد را با هتروزیگوت داسی مخلوط نمود. این روش الکتروفورز نیاز به تهیه همولیزات با غلظت هموگلوبین 10 گرم در دسیلیتر دارد و همولیزات را با سه روش زیر میتوان تهیه کرد:
- ابــتدا خـــــون بیمار را با نــــــرمال سیـــــــــلین 3 بار میشوییـــــــم و به یک حجم از گلبولهای قرمز فشرده شسته شـــــــده، 3 تا 4 حــــــــجم از محلــول ساپونین– سیانید (Saponin 100mg, KCN 50mg in 100ccW) میافزاییم.
- ابتدا نمونه بیمار را 5 دقیقه با دور g1200 سانترفوژ میکنیم. سپس 20 میکرولیتر از گلبول قرمز فشرده شده را با 150 میکرولیتر محلول همولیزات زیر مخلوط میکنیم:
Hemolysing reagent: 0/5 % Triton X – 100 in 100mg /L KCN
نمونه را به آرامی مخلوط میکنیم و 5 دقیقه صبر میکنیم.
- دو حجم از گلبول قرمز فشرده شسته شده را با یک حجم آب مقطر لیز میکنیم و سپس یک حجم تتراکلرید کربن (CCL4) به آن میافزاییم. نمونه را چند بار منجمد و ذوب میکنیم تا کاملاً لیز شود و سپس 2 حجم CCL4 با آن اضافه میکنیم. لوله را حدود 1 دقیقه تکان میدهیم و سپس 30 دقیقه با دور g1200 در درجه 4oC سانتریفوژ میکنیم. محلول رویی را به یک لوله تمیز منتقل کرده و میزان Hb را با آب مقطر حدود gr% 10 تنظیم مینماییم.
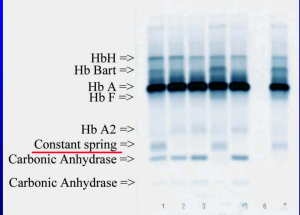
هموگلوبین کنستانت اسپیرینگ روی استات سلولز در PH قلیایی، بهویژه وقتی لکهگذاری غلیظ انجام شود، بین آنزیم کاربونیک انیدراز و هموگلوبین A2 قرار میگیرد؛ درحالیکه HbA¢2 که گونهای جهشیافته از زنجیرهی دلتاست، حرکت کندتری دارد و بین لکهگذاری و باند کاربونیک انیدراز قرار میگیرد.
هموگلوبین کنستانت اسپیرینگ با HPLC در دریچهی C جای میگیرد. هموگلوبینهای بارت و اچ در گروه هموگلوبینهای با حرکت سریع بوده و میتوان هموگلوبین اچ را با رنگآمیزی حیاتی، از قبیل کرزیل بلوی درخشان یا نیومتیلن بلو را بهصورت اچبادی یا مرفولوژی توپ گلف مشاهده کرد.
شکل زیر ترتیب قرار گرفتن هموگلوبینهای مختلف را با توجه به حرکت هموگلوبین A نشان میدهد. حرکت هموگلوبینها با توجه به حرکت A در دو گروه سریع و کند تقسیم میشوند. چنانچه باندهای الکتروفورز برای پروتئین رنگآمیزی شود شاهد باند کربنیک انیدراز در نزدیک محل لکهگذاری خواهیم بود و چنانچه از رنگآمیزی هیم (Heam) استفاده شود، باند فوق مشاهده نمیگردد.

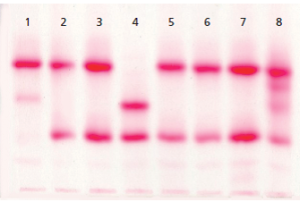
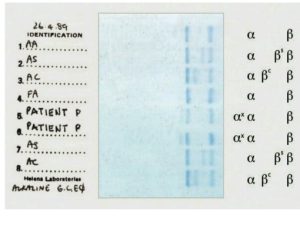
الکتروفورز هموگلوبین در بیمار شمارهی 1 روی استات سلولز الگوی A+S دارد؛ در این حالت چنانچه آزمایش حلالیت منفی باشد هتروزیگوت لپور باتوجه به پهنای واریان هموگلوبین که کمتر از 20 درصد است، مطرح میشود؛ البته ممکن است واریان G هم مطرح شود.
الکتروفورز هموگلوبین در بیمار شمارهی 4 الگوی C+S روی استات سلولز در PH قلیایی دارد؛ ولی ازآنجاییکه پهنای باند S و واریان هموگلوبین که در جایگاه C قرار گرفته است، مساوی است، هتروزیگوت دوبل داسی و C مطرح میگردد. واریانهای دیگر زنجیرهی بتا، یعنی هتروزیگوت دوبل C و D محتمل است.
توجه کنید که جایگاه D پنجاب و D ایران و S یکسان است. با آزمایش حلالیت میتوان هموگلوبین داسی را از این دو حالت افتراق داد. الگوی شمارهی 7 روی استات سلولز، هتروزیگوت هموگلوبینهای C یا E و یا O را نشان میدهد. البته پهنای باند E همیشه از C و O کمتر است.
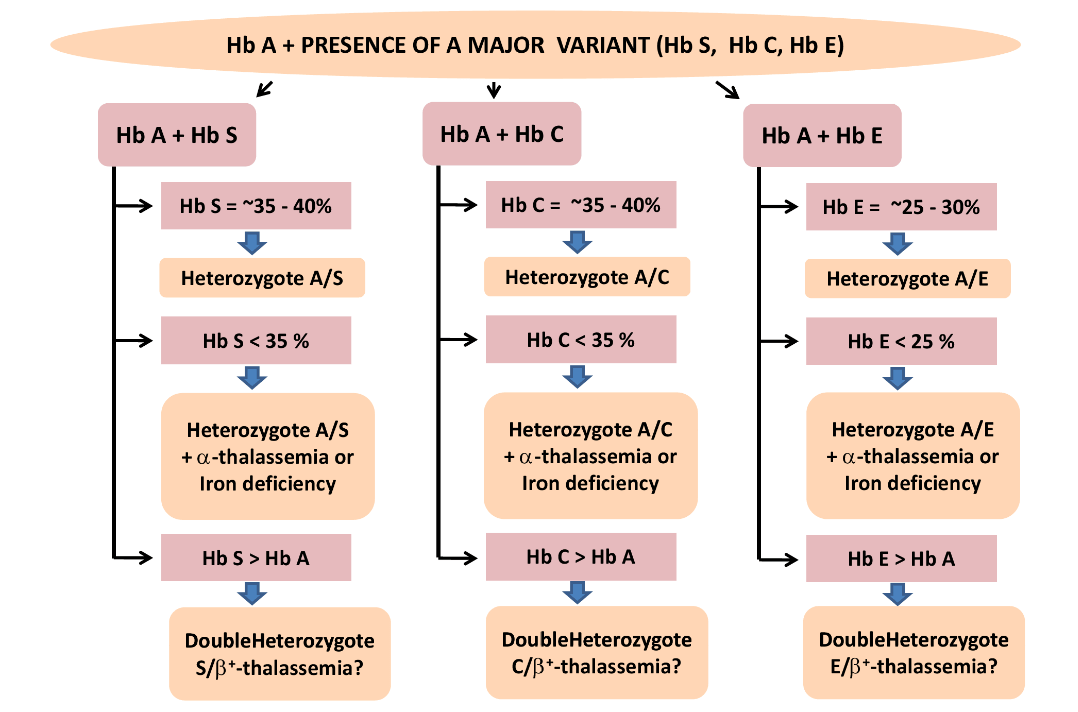
هموگلوبینهای همشارژ در یک گروه قرار میگیرند؛ برای مثال هموگلوبینهای A2 ,C ,O ,E در یک جایگاه قرار میگیرند ولی بر اساس پهنای باند ممکن است ایدههای تشخیصی مطرح شود، مثلاً باند A2 کمتر از ۷ تا ۸ درصد است. باندهای C و O در حالت هتروزیگوت بیشتر از ۳۵ تا ۴۰ درصد و باند E کمتر از ۳۰ درصد است.
باندهای S,D,G و لپور و آریا در یک جایگاه قرار میگیرند و در این حالت باند لپور و آریا کمتر از ۲۰ درصد و پهنای باندهای S و D در حالتهای هتروزیگوت بین ۳۵ تا ۴۰ درصد است.
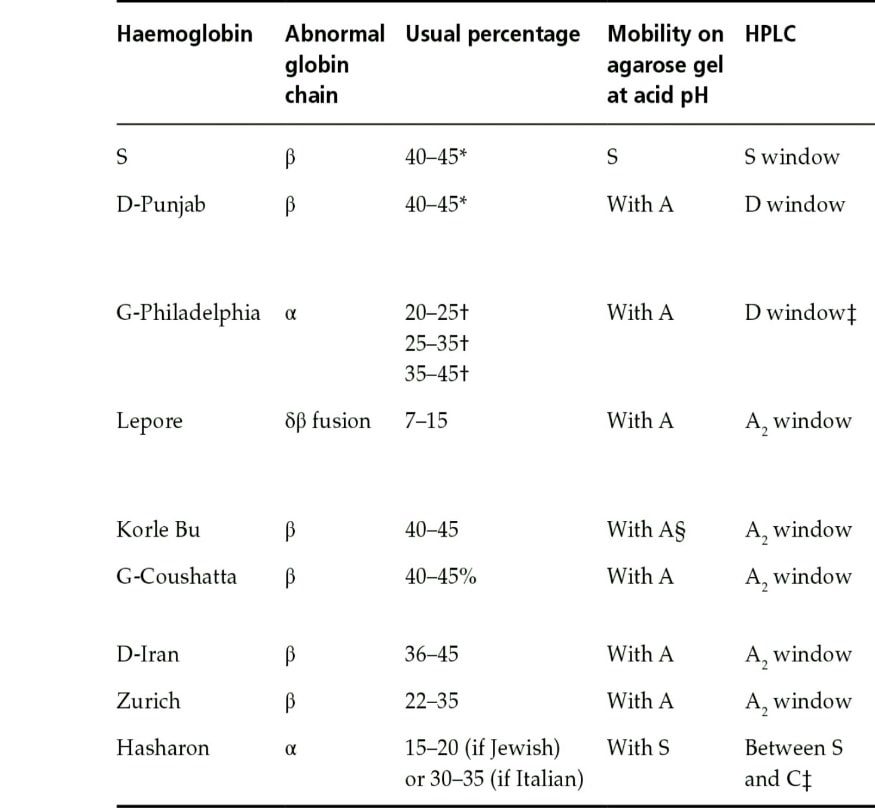
الکتروفورز هموگلوبین در بیمار شمارهی 2 و 3 تکباند S را روی استات سلولز نشان داده است. توجه کنید که همیشه باید با مشاهدهی تکباند S، الکتروفورز به شیوهی دیگری مانند ژل آگارز یا HPLC انجام شود؛ زیرا حالتهای هموزیگوت داسی (SS)، ترکیب داسی و تالاسمی (Sβ0)، هموزیگوتهای D و لپور، ترکیب داسی و هموگلوبینهای D و لپور ممکن است با تکباند S همراه با مقدار متغیر هموگلوبین F روی استات سلولز در PH قلیایی جلوه کنند.
در این حالت، چنانچه آزمایش حلالیت والدین مثبت باشد، هموزیگوت داسی مطرح است و چنانچه گلبولهای قرمز بیمار علاوه بر مورفولوژی داسی، مورفولوژی میکروسیت و هیپوکروم داشته باشد، احتمال الگوی ترکیبی داسی و تالاسمی مطرح است. گلبولهای قرمز در هموزیگوت داسی، نرموسیت و نرموکروم هستند. الگوی شمارهی 5 و 6 هموزیگوت داسی (SS) با ترانسفیوژن یا ترکیب داسی و تالاســمی (+S β) را با توجه به آزمایش مثبت حلالیت مطرح میکنند. تابلوی بالا دسته هموگلوبینهایی که همشارژ با هموگلوبین S روی استات سلولز هستند را نشان میدهد.
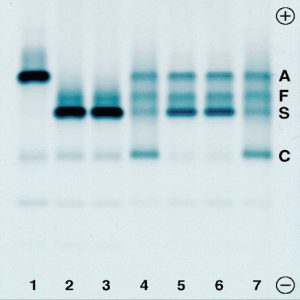
هموگلوبینهای اچ و بارت از هموگلوبینهای سریع هستند که با آزمایش اچ بادی یا توپ گلف که با رنگهای حیاتی انجام میشود میتوان هموگلوبین اچ را از بقیه گروه سریع (Fast) جدا کرد.
هموگلوبین اچ در رنگآمیزی حیاتی
برای مشاهده باندهای A’2 و کنستانت اسپرینگ که حرکت بسیار کندی دارند و در نزدیکی محل لکهگذاری و قبل از A2 قرار میگیرند بایستی از همولیزات نسبتاً غلیظ استفاده کرد تا پهنای باند آنها قابل مشاهده شود.
جداسازی رشتههای گلوبین و محاسبه نسبت سنتز زنجیره آلفا به بتا برای تشخیص موارد مشکلزای تالاسمی ماینور
با کروماتوگرافی ستونی کربوکسی متیل سلولز میتوان زنجیرهها را از هم جدا ساخت. برای جداسازی زنجیرهها از یکدیگر از بافر فسفات اورهدار (Urea phosphate) در PH=6-7 استفاده میشود. برای جداسازی بهتر و جلوگیری از ایجاد پیوندهای دیسولفیدی، یک احیاکننده مانند دای تیوتریتول (Di thio theritol) اضافه میشود.
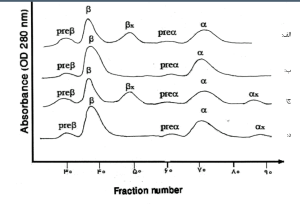
جداسازی زنجیرههای گلوبین با کروماتوگرافی توجه داشته باشید که زنجیره آلفا دارای شارژ مثبت و بتا منفی است. در گراف بالا به ترتیب یک بتای غیرطبیعی، حالت طبیعی، یک آلفا و یک بتای غیرطبیعی و یک آلفای غیرطبیعی مشاهده میشود.
با محاسبه نسبت سنتز زنجیره آلفا به بتا میتوان آن دسته از سندرمهای تالاسمی مینور بتا که با مقدار هموگلوبین A2 لب مرز 3/3 تا 3/5 قرار دارند را تشخیص داد و از طرف دیگر تشخیص جهشهای ظریف ژن بتا هم با این روش امکانپذیر است.
برای این کار سلولهای رتیکولوسیت از نمونه خون جدا میشود. یادآوری میشود که حدود ۲۰ درصد از سنتز هموگلوبین در مرحله رتیکولوسیتها انجام میگیرد، چون دارای RNA و ریبوزوم هستند. از گلبول قرمز بالغ نمیتوان استفاده کرد زیرا زنجیرههای اضافی هموگلوبین توسط آنزیمهای هیدرولیزکننده هضم شدهاند. برای مثال در تالاسمی مینور بتا مقداری زنجیره اضافی آلفا وجود دارد که در گلبول بالغ هضم میشوند و از این رو برای محاسبه سنتز از رتیکولوسیتها و یا ممکن است از گلبولهای قرمز هستهدار مغز استخوان استفاده شود که البته تهیه رتیکولوسیتها یک روش غیرتهاجمیتر است.
برای اندازهگیری نسبت سنتز زنجیرهها نخست رتیکولوسیتها با سانتریفوژ از گلبولهای بالغ جدا میشوند، لایه رتیکولوسیتها به علت وزن کمتر روی گلبولهای قرمز بالغ قرار میگیرند. با اضافه کردن آمینواسیدهای ضروری برای سنتز هموگلوبین، سنتز هموگلوبین در انکوباسیون ادامه مییابد و در این راستا یکی از اسیدآمینههای والین یا لوسین با کربن ۱۴ که اشعه بتا ساطع میکنند، نشاندار شدهاند.
پس از کروماتوگرافی و جدا شدن زنجیرهها، هر زنجیره به دستگاه بتا کانتر (β counter) داده شده و شــمارش اشعه در دقیقه (count per minute) محاسبه میشود. نسبت CPM آلفا به بتا برابر سرعت سنتز آلفا به بتا است. مقدار نسبت سنتز آلفا به بتا در حالت نرمال 1.05-0.93 است. در تالاسمی آلفا، بین 0.80-0.65 در کمبود یک ژن آلفا و بین 0.2 تا 0.3 در بیماری هموگلوبین اچ است. در تالاسمی مینور بتا این نسبت بین 2.22- 1.67 است.
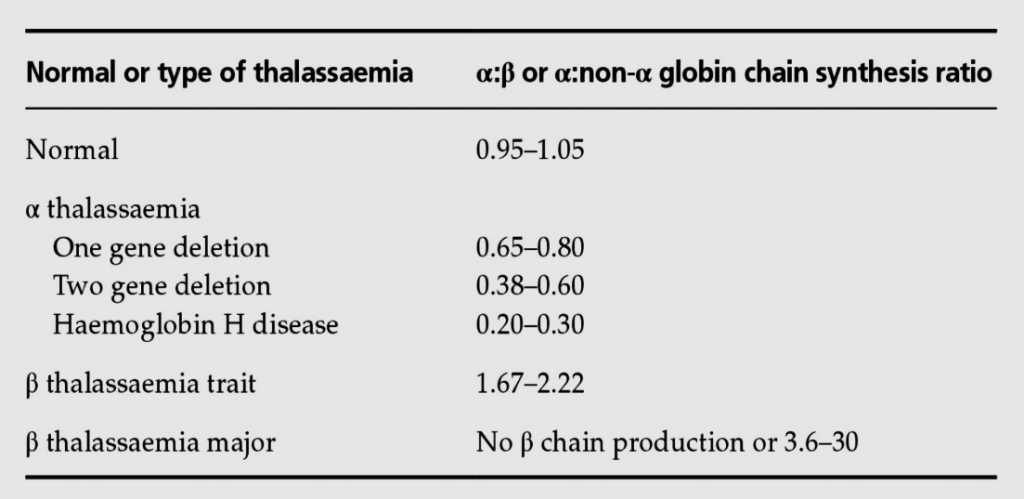
نسبت سنتز زنجیره آلفا به بتا در تالاسمیهای ماینور آلفا و بتا
تشخیص قطعی جهشها
چندین روش بر مبـــــــــــنای PCR برای تشخیص قطعی ناقلین تالاسمی فراهم شــــــــــــــده است؛ این روشها شــــــــــامل هیـــــــــبریدی شــــــدن با الیگــــــــــونوکلئوتیدهای اختصاصی آلل (Allele specific oligonucleotide hybridization) و آنالیز Gap-PCR ,ARMS-PCR و RDB و تعیین مستقیم توالی نوکلئوتیدها (Sequencing) است.
گفتنی است که با وجود بیش از 200 جهش شناختهشده تالاسمی بتا تنها تعداد محدودی از جهشها مسئول بیشتر از 90% تالاسمیها در یک منطقه جغرافیایی هستند و از این رو طراحی پرایمر (Primer) و پروب (Probe) که جهشهای یک منطقه را هدف قرار دهد، چندان مشکل نیست. آنالیز RAB از روشهای شایع برای تأیید جهشهای تالاسمی بتا است که پروبهای الیگونوکلئوتیدی مکمل آلل جهشیافته و نوع سالم آلل بر روی غشا قرار گرفته و با محصول PCR مجاور میگردند.
جهشهای ناشناخته با استفاده از تعیین توالی با استفاده از آمپلیکن (Amplicons) انجام میگیرد. برای شناسایی ژنهای حذفشده تالاسمی آلفا میتوان در صورت شناسایی نقطه شکســــــته از gap-PCR استفاده کرد و چنانچه تالاسمی آلفا بسیار مشکوک باشد ولی gap-PCR جواب قطعی به دست نداده است، استفاده از Real Time PCR ممکن است سودمند باشد. با این روش میتوان حذفهای آلفا که محل شکستگی واضحی ندارند و نیز هاپلوتایپهای سه آلفایی را تشخیص داد.
روش (Multiplex ligation probe amplification) MPLA قادر به آشکار کردن حذفهای ناشناخته چند صد جفت بازی و تعیین تعداد کپیهای ژنی برای مثال هاپلوتایپهای سه آلفایی است. برای شناسایی جهشهای نقطهای میتوان از آنالیز RDB و ARMS و تعیین سکانس مستقیم استفاده کرد.
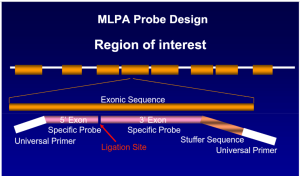
روش MLPA یک روش مولکولی است که برای تشخیص جهشهای طولی از قبیل حذفشدگیها و اضافهشدگیها و گاه جهشهای نقطهای و تعداد نسخههای ژن کاربرد دارد. روش MLPA بر پایه پروب (probe base) بوده و در یک واکنش MLPA میتوان 40 تا 50 منطقه از ژنوم را به طور همزمان از نظر تعداد نسخههای ژن (copy number) بررسی کرد. کیتهای غربالگری به منظور تشخیص بیماریهای مختلف با پروبهای مکمل سکانس ژنهای موتانت روی کروموزومهای مختلف و کیتهای اختصاصی برای یک جایگاه ژنی با پروبهای اختصاصی از قبیل تشخیص تالاسمیهای آلفا در دسترس است.
در روش MLPA پروبها، ژنوم مکمل را شناسایی کرده و پس از هیبرید شدن و لیگاسیون بجای ژنوم تکثیر میشوند. هر پروب دارای سه قسمت مجزا از سکانس برای هیبرید شدن با ژنوم و استافر (stuffer) و توالی برای اتصال به پرایمر است. پروبها پس از لیگاسیون توسط یک زوج پرایمر عمومی در صورت هیبرید شدن تکثیر میگردند. قطعه استافر هیچ شباهتی با مکمل سکانس ژنوم نداشته و برای داشتن فراورده با قطعات مختلف است.
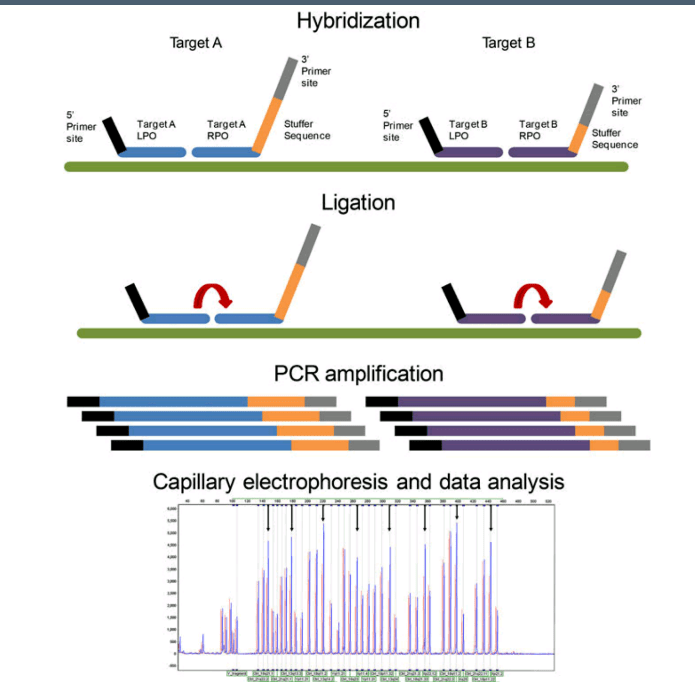
روش کار در MLPA
تالاسمیهای ماینور بتا
تاکنون بیش از 280 جهش نقطهای و حدود ۱۸ حذف بزرگ در ژن بتا در بروز تالاسمی شناخته شده است. جهشها گاهی موجب به صفر رساندن فرآورده ژن بتا (β0) گاهی با تولید اندک زنجیره (+β) و گاهی دارای جهش بسیار ظریف (++β) هستند. برخی از جهشهای ژن بتا تولید زنجیره بسیار ناپایدار بتا میکنند که حتی در حالت هتروزیگوت ایجاد علائم کمخونی و وابسته به تزریق خون میکند. هموگلوبینهای که با سرعت کم ساخته میشوند مانند هموگلوبین Eو کنستانت اسپرینگ و لپور در گروه تالاسمی جای میگیرند. هموگلوبین Malay حدود ۱۵ درصد ژن تالاسمی در مالزی و ۱۶ درصد موارد تالاسمی در جنوب تایلند و چین و اندونزی را در بر میگیرد. هموگلوبین مالای در الکتروفورز و کروماتوگرافی حرکتی شبیه به هموگلوبین A دارد.
گاهی ژن بتا سالم است ولی جهش در فاکتورهای خارج از ژن مانند ERCC2و GATA1 و ASH1L باعث بروز تالاسمی میشوند. جهش ژن GATA1 علاوه بر تالاسمی موجب ترومبوسیتوپنی هم میشود. هتروزیگوت بتا دارای ژنوتایپهای مختلفی است که شایعترین آنها β0β و β+β میباشد.
تالاسمی مینور بتا رلهای محافظتی از قبیل کاهش سکته قلبی، شیوع کمتر فشارخون، کاهش سکته مغزی، متعادل بودن غلظت کلسترول و LDL و رل محافظتی در برابر مالاریا دارد.
بنا به مطالعات گروه سیسیلی در ناقلین ماینور خستگی، شانس بیشتر روماتیسم مفصلی، شیوع بیشتر نقص لوله عصبی، حجم بیشتر طحال، استعداد به سنگ کیسه صفرا بهویژه با به ارث بردن سندرم گیلبرت، اختلال عملکردی لولههای کلیوی، هموکروماتوز، هپاتیت C با عارضه فیبروز و رسوب بیشتر آهن با شیوع بیشتر در تعدادی از ناقلین مشاهده شده است.
جداسازی حالتهای β0/β از β+/β با اندکسهای خون و اندازهگیری هموگلوبین A2 همپوشی دارند ولی چنانچه MCV کمتر از ۶۷ و MCH کمتر از ۲۱ باشد احتمال β0/β را مطرح میکند. در تالاسمی ماینور بتا تنها یک ژن بتا نقص β0 (بدون فراورده) یا +β (5 تا 30 درصد فعالیت نرمال) دارد و از این رو، ژنوتایپ را میتوان بهصورت ββ0 و +ββ نمایش داد. دادههای CBC در تالاسمی ماینور بتا افزایش تعداد گلبولهای قرمز (5 تا 7 میلیون)، کاهش MCV (غالباً کمتر از ۷۲)، کاهش MCH (کمتر از ۲۲) و مقدار نرمال یا کاهش MCHC و افزایش دو برابری رتیکولوسیت را نشان میدهد. گسترهی محیطی میکروسیت و هایپوکروم یکدست، انکلوزیون بازوفیلیک استیپلینگ و تعدادی تارگت سل را در زمینهی شلوغ گلبولهای قرمز نشان میدهد.
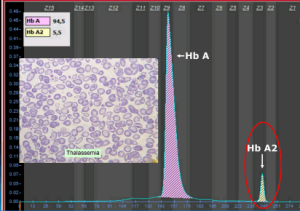
گسترهی خون محیطی در سندرمهای تالاسمی مینور، زمینهی شلوغ گلبولهای قرمز میکروسیتیک هایپوکروم همراه با مقداری تارگت سل و انکلوزیون بازوفیلیک استیپلینگ را نشان میدهد .افزایش بیشتر از 3/3 در صد از HbA2 ارزش بالینی دارد مقدار نرمال آن 2 تا 3/3 در صد است.
گونههایی از تالاسمی ماینور بتا با اندکسهای نزدیک به نرمال همراه با افزایش HbA2 است؛ که برای مثال میتوان به همراهی تالاسمی ماینور آلفا با فقدان یک تا دو ژن آلفا، با تالاسمی ماینور بتا اشاره کرد. چنانچه سندرمهای تالاسمی ماینور کنار گذاشته شود، افزایش HbA2 ممکن است ناشی از کمخونی مگالوبلاستیک، کمخونیهای دیس اریتروپوئتیک، پرکاری تیروئید، هموگلوبینهای ناپایدار، بیماری ویلسون و مصرف داروهای ضدویروسی باشد. گفتنی است همراه شدن سندرمهای تالاسمی ماینور با کمخونی مگالوبلاستیک یا بیماریهای کبدی ممکن است اندکسها را نرمال کند.
میزان MCV در خانمهای حامله با تالاسمی ماینور حدود 2 فمتولیتر و در خانمهای حامله بدون تالاسمی حدود 4 فمتولیتر بالا میرود. برای جداسازی آنمی فقر آهن از سندرمهای تالاسمی ماینور که هر دو تصویری میکروسیت و هیپوکروم دارند فرمولهای متعددی ارائه شده است ولی شاید میزان MCV از همه مهمتر باشد. اصطلاح میکروسیت و هایپوکروم به MCV≤80 و MCH<27 اطلاق میشود. مرز مشخصی از کاهش MCV برای جداسازی سندرمهای تالاسمی ماینور از آنمی فقر آهن وجود ندارد؛ ولی بیشتر مطالعات MCV کمتر از ۷۲ را برای سندرمهای تالاسمی ارزشمند میدانند و از این رو بهتر است پیش از آهندرمانی برای شخصی که MCV کمتر از ۷۲ دارد برای تشخیص سریعتر، روشهای مولکولی از قبیل تعیین نسبت سنتز زنجیرهی آلفا به بتا را مدنظر قرار داد بدین مفهوم که چنانچه در فرد بالغ مقدار MCV کمتر از ۷۲ باشد بهتر است بهجای آهن درمانی اقدام به آزمایشهای مولکولی کرد و این در صورتی است که آزمایشهای روتین مانند اندازهگیری هموگلوبین A2 کمکی نکرده باشد.
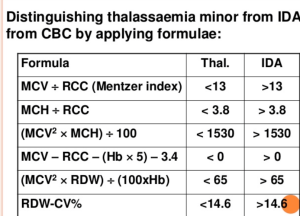
فرمولهای افتراق دهنده تالاسمیهای ماینور از فقر آهن. دادههای میکروسیت و هایپوکروم در فرمولهای فوق قرار میگیرد
چنانچه بیماریهای کبد و یا کمخونی مگالوبلاستیک با تالاسمی ماینور همراه شود امکان دارد مقادیر MCVو MCH در طیف طبیعی قرار گیرد گرچه هموگلوبین A2 افزایش دارد. داروهای زیدوودین (Zidovudin) و هیدروکسی کاربامید اثری مشابه فوق بر اندکسهای خون دارند.
شخص مبتلا به تالاسمی ماینور که بر اثر یک بیماری دیگر اقدام به جراحی و برداشتن طحال کرده است دارای تصویری ناهنجارتر، از نظر مورفولوژی گلبولهای قرمز میگردد.
در افتراق آنمی فقر آهن از تالاسمی ماینور بتا تعداد گلبولهای قرمز بیشتر از 5/5 میلیون، حضور بازوفیلیک استیپلینگ به نفع تالاسمی و کاهش کمتر از ۵ میلیون گلبول قرمز و حضور الیپتوسیتهای کمرنگ مدادی شکل احتمال فقر آهن را مطرح میکند. آنمی فقر آهن شدید موجب کاهش هموگلوبینهایی، مانند HbH و HbA2 میشود و از این رو آن دسته از ناقلان تالاسمی ماینور را که افزایش HbA2 در آنها اندک است و بین 3/5 تا 4 درصد است، در طیف طبیعی میآورد که پس از درمان آهن باید آزمایش CBC و HbA2 تکرار کرد.
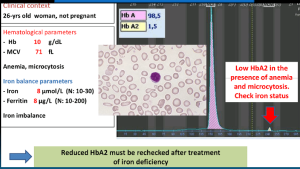
سطح هموگلوبین A2 زیر 2 در صد همراه با میکروسیت و هایپوکروم در بیماری هموگلوبین H در فقر آهن پیشرفته و در تالاسمی ماینور بتا در همراهی با تالاسمی دلتا مشاهده میشود. چنانچه اندکسها نرمال باشد ممکن است مطرح کننده تالاسمی دلتا باشد.
در اسکرین تالاسمی به نظر میرسد که اندکس MCH<27 بهتر از MCV<80 باشد زیرا پارامتر MCV تحت اثر مانده شدن خون، دیابت و کمآبی بدن، لکوسیتوز و کافی نبودن شستشوی دستگاه قرار میگیرد. چنانچه روزنه سلول شمار بهتدریج با لاشههای سلولی گرفته شود و روزنه آن تنگ شود با افزایش تدریجی کاذب MCV روبهرو میشویم. دیابت و کم آب بودن بدن موجب تورم گلبولهای قرمز در محلول شمارش دستگاه و افزایش کاذب MCV میگردند.
همیشه وقتی با اندازهگیری هموگلوبین A2 به دنبال تشخیص تالاسمی ماینور بتا هستید باندهای دوبخشی (Split band) هموگلوبین A2را مدنظر داشته باشید. باندهای دوبخشی هموگلوبین A2 در وراثت واریانتهای آلفا و واریانتهای زنجیره دلتا به ارث میرسند. برای مثال واریان زنجیره دلتا ایجاد هموگلوبین A’2 و واریانت آلفا با زنجیره دلتا ایجاد باندهای باریک میکند. برای مثال واریان αG ایجاد هموگلوبین Hb G2 میکند. در این موارد کل هموگــلوبین A2 برابر با A2+ A’2 یا برابر A2+ G2 و A2 variant+ A2 میباشد.
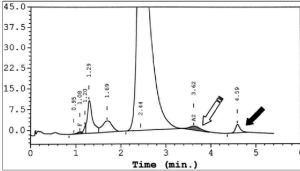
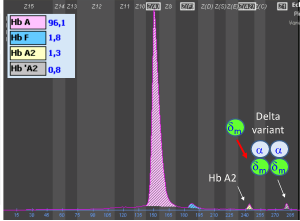
توجه کنید که HbA2 در فرد ممکن است به علت واریانهای زنجیرهی آلفا یا دلتا، دوبخشی باشد؛ برای مثال هموگلوبینهای G و آریا که واریان آلفا هستند، چنانچه در کنار آلفای سالم قرار گیرند هموگلوبین دوبخشی A2 را ایجاد میکند. زنجیره آلفای سالم و دلتا هموگلوبین A2 را پدید میآورد و آلفای جهشدار با دلتا واریان دیگر A2 را شکل میدهد که در الکتروفورز در یک مکان قرار نمیگیرند و HbA2 بیمار مجموع دوبخشی است. فلش سفید هموگلوبین A2 و فلش مشکی واریان A‘2(HbB2) را با HPLC نشان میدهد.
هموگلوبین A‘2(HbB2) نوعی هموگلوبین A2 با زنجیرهی جهشدار دلتاست که در الکتروفورز روی استات سلولز، کندتر از A2 حرکت میکند و در حضور یک دلتای سالم، باند دوبخشی ایجاد میکند. روش ستونی اندازهگیری HbA2 هر دو را اندازهگیری میکند؛ ولی در الکتروفورز در جایگاههای مختلف قرار میگیرد. در این موارد کل هموگلوبین A2 بـــــــرابر با A2+ A’2 یا برابر A2+ G2 و A2 variant+ A2 میباشد. هموگلوبین A¢2 روی استات سلولز در PH قلیایی کندتر از A2 و نزدیک به باند انیدراز کربونیک قرار میگیرد. باندA¢2 در HPLC در دریچهی S قرار میگیرد.
مشاهده واریانتهای A2 با الکتروفورز روی استات سلولز مشکل است و نیاز به لکهگذاری غلیظ دارد ولی الکتروفورز با روشهای کاپیلاری و HPLC میتوان با سهولت بیشتری آنها را جستجو کرد. اهمیت باند دوبخشی A2 نهتنها در تشخیص تالاسمی مینور بتا اهمیت دارد بلکه برای شناسایی واریانتهای آلفا هم اهمیت دارد.
هموگلوبین A’2 در HPLC در دریچه S و با الکتروفورز کاپیلاری در ناحیه ۱ و ۲ ظاهر میشود. برخی از افراد ممکن است برای A’2 هموزیگوت باشند و مقدار هموگلوبین A2 صفر باشد در همراهی تالاسمی ماینور بتا با هموزیگوت 0 δ نیز مقدار هموگلوبین A2 را صفر میکند.
جهشهای ناحیه پروموتور ژن بتا مانند 88C>T- و 29A>G – و نیز حذف 5’ ژن بتا و جهش در رمز آغازین با سطح A2 بالاتر همراه است. مقدار معمول A2 در هتروزیگوت بتا معمولاً ۴ تا ۵ درصد است. زمانی که مقدار هموگلوبین A2 بالا بود نیازی به انجام Hb F نیست ولی چنانچه نرمال باشد برای کنار گذاشتن تالاسمی F یا تالاسمی هتروزیگوت دلتا بتا یا سندرم پابرجایی سنتز F (HPFH) اندازهگیری هموگلوبین F سفارش میشود.
الکتروفورز هموگلوبین نیز برای جستجوی هموگلوبین لپور توصیه میشود. جهشهای غیرفعالکننده فاکتور KLF1 با MCV حدود 68 و MCH حدود 21 و افزایش هموگلوبین A2 تا 4/9 درصد و افزایش هموگلوبین F همراه است. همراهی جهش KLF1 با تالاسمی هتروزیگوت بتا آن را شبیه به تالاسمی اینترمدیا در میآورد. همچنین جهشهای هموزیگوت KLF1 با تصویر تالاسمی بتای اینترمدیا ظاهر میشود. تشخیص تالاسمی ماینور بتا در نوزاد مشکل است ولی در ششماهگی همپوشی مقدار A2 با نوزاد ناقل دیده نمیشود و سطح A2 بالا است.
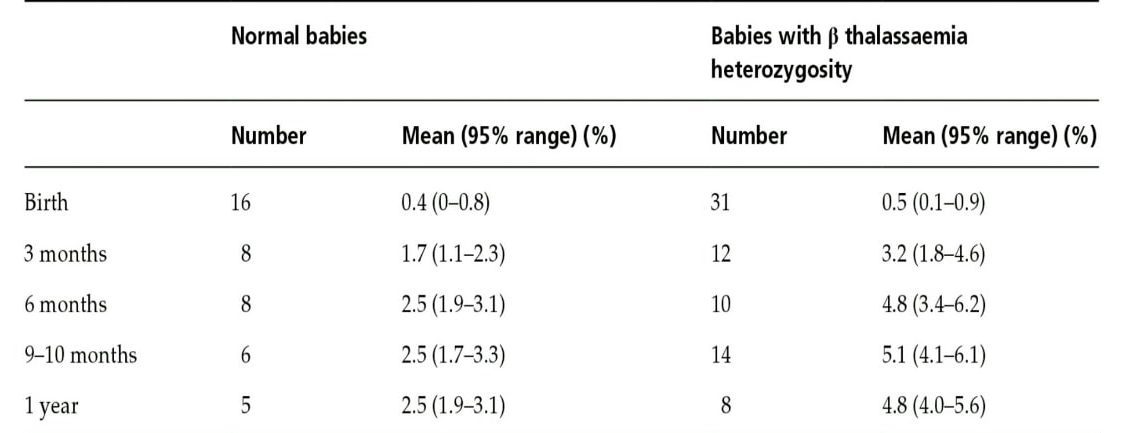
هموگلوبین A2 در بدو تولد 0.2 تا 0.3 درصد است و در یک تا دوسالگی بهاندازهی هموگلوبین بالغین میرسد. شیب افزایش آن در سال اول زندگی است و تا دوسالگی افزایش کندتری را نشان میدهد. گفتنی است سنتز رشتههای آلفا و بتا در پیشسازهای گلبول قرمز و در رتیکولوسیتها هم ادامه دارد؛ درحالیکه سنتز زنجیرهی دلتا قبل از مرحلهی رتیکولوسیت اتمام میپذیرد. هموگلوبین A2 ازنظر کارایی شبیه هموگلوبین A است. در ناقلین تالاسمی ماینور بتا افزایش سطح هموگلوبین A2 از 6 ماهگی مشاهده میشود
مواردی وجود دارد تحت عنوان تالاسمی ماینور خاموش که نهتنها اندکسها بلکه Hb A2 طبیعی است. این موارد در همراهی با ماینور بتا چنانچه به ارث رسیده شوند ممکن است با تالاسمی اینترمدیا بروز کنند که شایعترین آنها جهشهای 101C>T- و 92C>T- میباشد.
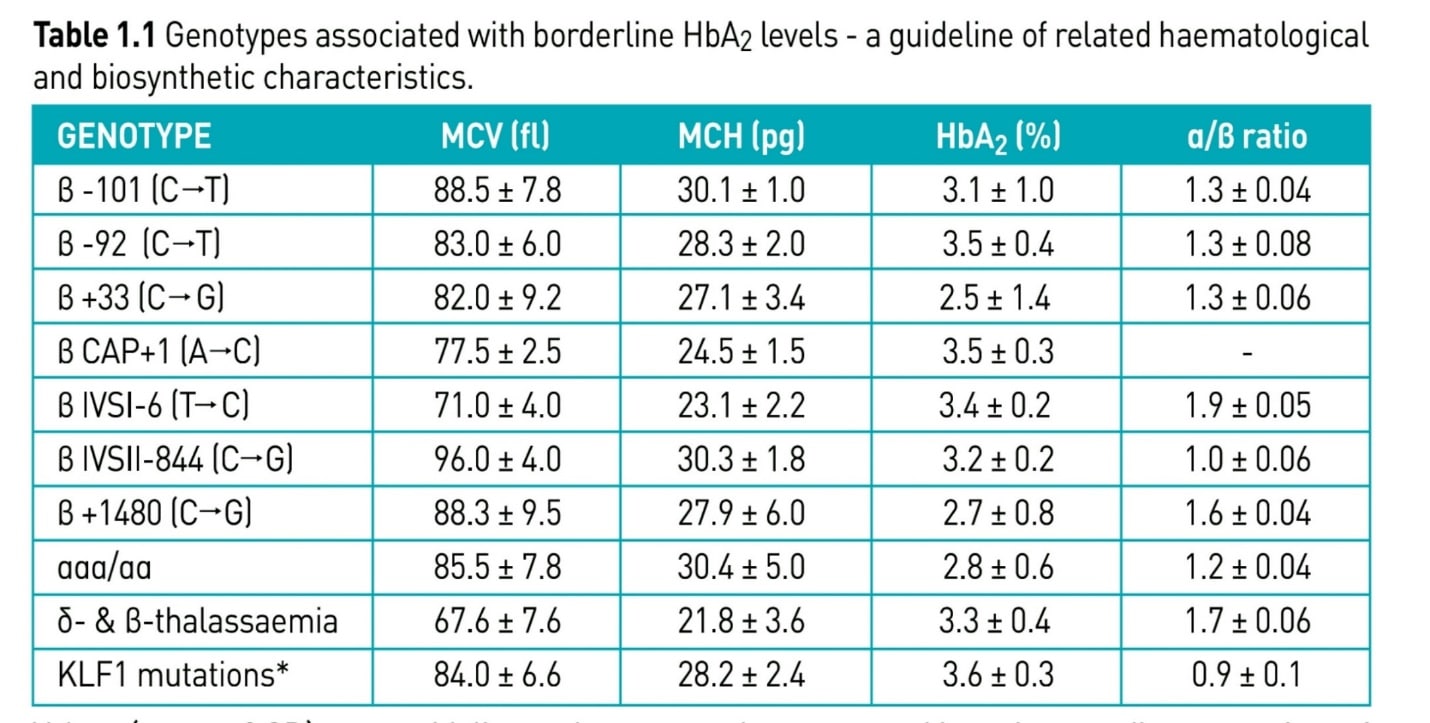
جهشهای ژن بتا که سطح هموگلوبین A2 لب مرز نرمال یا طبیعی است و اندکس خون هم ممکن است لب مرز نرمال باشد
آنمی فقر آهن موجب کاهش 0/5 درصدی هموگلوبین A2 میگردد و مشکل تشخیصی برای آن دسته از افراد مبتلا به تالاسمی ماینور که هموگلوبین A2لب مرز بالا مانند 3/8 تا 4 دارند ایجاد کرده و آن را در مرز طبیعی قرار میدهد. نکته جالب اینکه کمبود اسیدفولیک در شخص مبتلا به تالاسمی ماینور بتا موجب کاهش هموگلوبین A2 میگردد ولی در افرادی که تالاسمی ماینور بتا ندارند کمخونی مگالوبلاستیک ناشی از کمبود ویتامین B12 و اسیدفولیک باعث افزایش هموگلوبین A2 میگردد.
وراثت همزمان تالاسمی آلفا (کمبود دو ژن آلفا) با هتروزیگوت تالاسمی بتا ممکن است اندکسها را در مرز طبیعی آورد ولی هنوز ممکن است که هموگلوبین A2 بالا باشد.
پلیسایتمی ورا با اندکسهای تالاسمی
در پلیسایتمی ورا ذخایر آهن مغز استخوان به صفر نزدیک میشود. از این رو افزایش شمارش گلبولهای قرمز با بیش از 6 میلیون بهصورت میکروسیت و هایپوکروم در میآید که با نمای تالاسمی ماینور اشتباه میشود. گفتنی است که در پلیسایتمی ورا برخلاف
تالاسمی ماینور در اکثر موارد پانمایلوز با افزایش هر سه رده سلولی قرمز و سفید و پلاکت همراه است. در تالاسمی ماینور بهندرت هموگلوبین بیشتر از 14 میشود

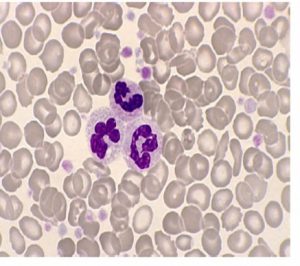
گسترهی محیطی بیمار مبتلا به پلیسایتمی ورا در مرحلهی اریتروسایتوتیک که با انبوه گلبولهای قرمز میکروسیت و هایپوکروم و پانمایلوز همراه است و کاهش اندکسها ناشی از کمبود شدید آهن است. آزمایش جهش Jak2 در بیمار فوق مثبت و هموگلوبین A2 برابر 2/3 و نسبت سنتز زنجیره آلفا به بتا حدود یک و حذف یا جهشی در ژنهای آلفا و بتا مشاهده نشده است.
همراهی ژنوتیپ اچ با ماینور بتا شبیه تالاسمی اینترمدیا با میکروسیتوز بسیار شدید با میانگین MCV حدود 55 و MCH حدود 15 و پوییکیلوسیتوز جلوه میکند و مقدار هموگلوبین A2 در این موارد طبیعی یا لب مرز 3/4 تا 3/7 است.
بیماریهای کبد با انباشت چربی روی غشای گلبول قرمز موجب طبیعی شدن اندکسها شده درحالیکه هموگلوبین A2 همچنان بالا باقی میماند.
وراثت هتروزیگوت بتا با ژنهای سهتایی آلفا مانند ααα/αα یا ααα/ααα موجب رسوب زنجیرههای اضافی آلفا و تصویر تالاسمی اینترمدیا میگردد. وراثت زنجیره بتای ناپایدار حتی در حالت هتروزیگوت علائم دار و وابسته به تزریق خون میگردد که در این حالت زنجیره ناپایدار همراه با زنجیره آلفا بهصورت انکلوزیون در گلبول قرمز رسوب میکند؛ این حالت عنوان تالاسمی ماینور غالب (Dominant) یاد میشود.
گفتنی است برخی جهشهای تالاسمی ماینور بتا با علائم بالینی، مانند بزرگ شدن طحال و ژاندیس و افزایش شیوع سنگ کیسهی صفرا و کمخونی همراه است که با عنوان تالاسمی ماینور غالب بتا شناخته میشود. در این نوع تالاسمی شدت کمخونی شبیه تالاسمی اینترمدیاست و ممکن است تزریق خون، ضروری باشد. خون محیطی انکلوزیون بازوفیلیک استیپلینگ و گلبول قرمز هستهدار را نشان میدهد. انکلوزیونهای اریتروئیدی از زنجیرهی آلفا و بتای ناپایدار شکل میگیرند؛ درحالیکه در تالاسمی ماژور بتا تنها، از اضافههای زنجیرهی آلفا درست شدهاند. تالاسمی ماینور غالب درنتیجهی جهش در ناحیهی 3¢ قسمت 1:3 انتهایی اگزون 2 یا اگزون 3 ژن بتاست. این جهشها با تولید زنجیرهی ناپایدار طویلشده یا کوتاهشدهی β همراه است که موجب راسب شدن آن، به همراه زنجیرههای آلفای سالم میشود و انکلوزیونها موجب آسیب غشا و خونسازی بیهوده میشود.
جهشهای خارج ژنی که منجر به تالاسمی میگردد:
جهشها و پلیمورفیسمهای خارج از خوشه ژنهای آلفا و بتا که منجر به سنتز غیرطبیعی زنجیرهها یا فنوتایپ تالاسمی میگردند عبارتند از
- جهش در ژن ATRX روی کروموزوم ایکس که فرآورده طبیعی آن بیان ژن آلفا را کنترل میکند. جهش در این ژن با بیماری هموگلوبین اچ با اختلالات اسکلتی بهویژه چهره دیسمورفیسم و اختلال یادگیری و عقبماندگی رشد ذهنی همراه میباشد.
- جهش در ژن FCP1 روی کروموزوم ایکس با تداوم سنتز هموگلوبین F همراهی دارد.
- پلیمورفیسم داخل ژنی HBSIL-MYB روی کروموزوم ۶ با تداوم سنتز هموگلوبین F همراهی دارد.
- جـــهش در ژن ERCC2(XPD) روی کـــرومــــوزوم ۱۹ بــــا تریــــکوتیـــودیستروفی (TRICHO THIO DYSTROPHY) و تالاسمی بتا همراهی میشود.
- جهش ژن GATA1 روی کروموزوم ایکس با ترومبوسیتوپنی و تالاسمی بتا وابسته به X همراه است.
- غیرفعال شدن ژن BCL11A روی یک هاپلوتیپ با افزایش هموگلوبین F و اختلالات در درک مفاهیم و چهره دیسمورف همراهی دارد.
- جهش در ژن KLF1 روی کروموزوم ۱۹ با افزایش هموگلوبین A2 همراهی دارد.
- جهش در ژن ASH1L روی کروموزوم یک با تالاسمی بتا همراهی دارد.
هموگلوبین لپور
تقاطع نابرابر در میوز با حذف قسمت 3¢ ژن دلتا و 5¢ ژن بتا منجر به ادغام قسمتی از ژنهای دلتا و بتا میشود. ژنهای ادغامشده تحت اثر پروموتور ضعیف ژن دلتا قرار گرفته و از این رو، با سرعت بسیار کمتری در مقایسه با زنجیرهی بتا ساخته میشود و در گروه هموگلوبینوپاتی تالاسمی قرار میگیرد. به این واریان، هموگلوبین لپور گفته میشود که میتوان فرمول آن را بهصورت α2(dβ)2 نمایش داد.
با توجه به محل ادغام ژنی، واریانهای مختلف لپور تولید میشود که شایعترین آنها لپور بوستون یا واشنگتن است. لپور بالتیمور و هلندیا از انواع دیگر لپور است. گسترهی محیطی در هتروزیگوت لپور شبیه تالاسمی ماینور است و الکتروفورز، 5 تا 15 درصد باند لپور همراه با کاهش HbA2 و اندکی افزایش HbF را نشان میدهد. هموگلوبین لپور روی استات سلولز در PH قلیایی همگام با هموگلوبین S و روی ژل آگارز در PH اسیدی با هموگلوبین A همگام است و در HPLC در جایگاه A2 قرار میگیرد.
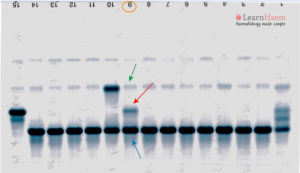
هموگلوبین لپور روی استات سلولز در جایگاه هموگلوبینهای S,D,G و با الکتروفورز به روش کاپیلاری در Zone D قرار میگیرد و با در نظر گرفتن پهنای باند که کمتر از 20 در صد است ممکن است بتوان از بقیه جدا کرد. فلش قرمز به هموگلوبین لپور اشاره دارد.
هموزیگوت لپور شبیه تالاسمی ماژور یا اینترمدیاست و الکتروفورز، تنها هموگلوبینهای F و لپور را نشان میدهد. هتروزیگوت ترکیبی لپور و تالاسمی نیز شبیه تالاسمی ماژور یا اینترمدیا جلوه میکند. گفتنی است کاهش سرعت سنتز هموگلوبین E نیز آن را در گروه هموگلوبینوپاتیهای تالاسمی میآورد. هتروزیگوت ترکیبی E/β0 شبیه تالاسمی ماژور جلوه میکند.
تالاسمی دلتا بتا
در تالاسمی دلتاـبتا ژنهای معیوب دلتا و بتا روی یک هاپلوتایپ کروموزوم ۱۱ مشاهده میشود و هاپلوتایپ دیگر، دربردارندهی ژنهای سالم دلتا و بتاست. تالاسمی ماینور دلتاـبتا ( δβ)/ δβ0 به نام تالاسمی F نیز شناخته میشود؛ زیرا ژن گاما بهطور جبرانی افزایش بیان دارد و میزان هموگلوبین F به 5 تا 20 درصد میرسد؛ بنابراین حضور اندکسهای تالاسمیک با افزایش HbF و مقدار نرمال تا کاهش یافته HbA2 (میانگین 2/4) تشخیص را بهسوی تالاسمی هتروزیگوت دلتاـبتا میبرد. تالاسمی هتروزیگوت دلتاـبتا ازنظر بالینی خفیف است و هموگلوبین، کاهش اندک دارد. مقدار MCV 65 تا 79 و MCH 21 تا 26، متغیر است. گفتنی است تالاسمی هموزیگوت دلتاـبتا نیز آرام است و بیشتر هموگلوبین از HbF (تقریباً 100 درصد)، تشکیل شده است.
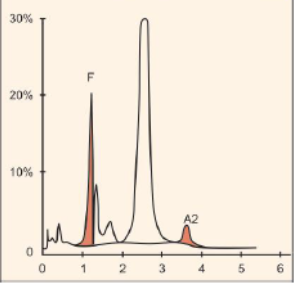
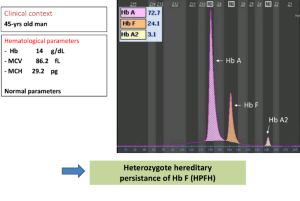
در تالاسمی هتروزیگوت دلتاـبتا که به آن تالاسمی F گفته میشود، هرچند ژنهای دلتا و بتا حذف میشود یا عملکردشان کاهش مییابد، کاهش اندکسها با افزایش 5 تا 15 درصدی هموگلوبین F با پخش غیریکنواخت و بیتعادلی زنجیرههای آلفا و غیرآلفا مشخص میشود. پخش پانسلولار HbF در تداوم سنتز هموگلوبین F ممکن است حذفی یا غیرحذفی باشد. حذفی، به مفهوم حذف شدن ژنهای دلتا و بتا در کنار ژن گاماست که در این حالت، ژن گاما تقریباً کمبود زنجیرههای بتا و دلتا را جبران میکند. حالت هتروزیگوت HPFH با 15 تا 30 درصد هموگلوبین F و یک تا دو درصد هموگلوبین A2 و اندکسهای نرمال یا نزدیک نرمال ظاهر میشود. هموزیگوت آن با 100 درصد هموگلوبین F همراه است.
در تالاسمی دلتا (+δ0 or d) سنتز زنجیرههای دلتا، صفر یا نزدیک به صفر میشود. تالاسمی دلتا هیچگونه علامت بالینی و آزمایشگاهی ندارد؛ ولی همراهی تالاسمی دلتا با تالاسمی ماینور بتا، موجب کاهش HbA2 میشود و از این رو، مشکلات تشخیصی ایجاد میکند.
ممکن است بتوان با مطالعهی اعضای خانواده، به همراهی تالاسمی دلتا با تالاسمی ماینور بتا که سبب کاهش HbA2 شده است، پی برد که در این حالت، ممکن است اعضای سالم خانواده با اندکسهای کاملاً طبیعی، کاهش HbA2 را نشان دهند.
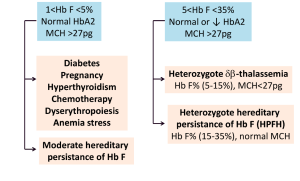
موارد مهم افزایش هموگلوبین F بین یک تا 5 در صد و بین 5 تا 35 در صد
در تالاسمی هتروزیگوت بتا در حالت β0β و β+β گستره محیطی نمای میکروسیت و هایپوکروم یکدست با تعدادی تارگت سل نشان میدهد ولی چنانچه تالاسمی +β یا β0 با ژنهای D پنجاب یا O عرب همراه شود علاوه بر نمای میکروسیت هایپوکروم تعداد زیادی تارگت سل و نیز تعدادی گلبولهای فشرده با لبههای نامنظم و اسفروسیت مشاهده میگردد که در این موارد بایستی شک را بهصورت هتروزیگوتهای دوبل تالاسمی و هموگلوبینهای D و O عرب و C برد.
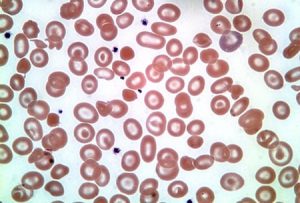
نمای میکروسیت هایپوکروم تعداد زیادی تارگت سل و نیز تعدادی گلبولهای فشرده با لبههای نامنظم و اسفروسیت شک را بهسوی هتروزیگوتهای دوبل تالاسمی و واریانهای بتا میبرد.
سندرمهای تالاسمی آلفا
تالاسمیهای آلفا ناشی از حذف یا جهش ژنهای آلفا است. ژنوتیپ آلفای یک شخص سالم αα/αα است ولی در واقع بهصورت α1α2 / α1α2 است.
ژنهای آلفا ۲ حدود ۷۵ درصد سنتز زنجیره آلفا را به عهده دارند و از این رو حذف یا جهش آنها میتواند منجر به کمخونی شود. حذف یک آلفا دارای علائم بالینی و هماتولوژی نیست و میزان MCV بین ۷۵ تا ۸۵ متغیر و با حالت نرمال همپوشی دارد. در بدو تولد تا ۳ الی ۶ ماهگی ممکن است بتوان یک تا سه درصد هموگلوبین بارت (γ4) را در الکتروفورز مشاهده کرد، البته در نوزاد نرمال هم ممکن است بین نیم تا یک درصد مشاهده شود.
فقدان دو ژن آلفا بهصورت حذف سیس αα/– یا ترانس α/-α- با هموگلوبین بارت بین ۳ تا ۸ درصد در بدو تولد همراه است. میزان هموگلوبین بارت در فنوتایپ حذفی سیس بیشتر از ترانس است. میزان MCV نرمال نوزاد بین ۱۰۵ تا ۱۲۰ فمتولیتر است ولی در این موارد اغلب کمتر از ۹۴ است. در بزرگسالان تالاسمی ماینور آلفا با حذف دو ژن آلفا بهصورت میکروسیتیک و هایپوکروم و نرمال بودن هموگلوبین A2 جلوه میکند و تنها با روشهای مولکولی مانند Gap PCR و یا محاسبه نسبت سنتز زنجیره آلفا به بتا (α/β ratio) میتوان آن را تشخیص داد.
در بزرگسالان، کاهش اندکسهای خون همراه با HbA2 کمتر از 2/5 درصد به شرطی که آنمی فقر آهن رد شده باشد، احتمال تالاسمی ماینور آلفا مطرح میشود؛ از آنجا که جهشهای ظریف تالاسمی ماینور بتا با کاهش اندکسها و مقدار نرمال HbA2 با تالاسمی ماینور آلفا نمایی یکسان دارد، آزمایش نسبت سنتز زنجیرهها یا روشهای مولکولی دیگر توصیه میشود. ممکن است بتوان با دقت فراوان هموگلوبین H(β4) را در حالت (–/αα) در تعداد بسیار کمی از گلبولهای قرمز با انکوباسیون خون با رنگهای حیاتی تشخیص داد و چنانچه، نخست خون بیمار را سانتریفیوژ کنید و از لایهی زیر بافی کوت، یک قطره خون برای رنگآمیزی اچبادی بردارید، احتمال مشاهدهی اچبادی افزایش مییابد؛ زیرا این قطره، غنی از رتیکولوسیت است و هنوز زنجیرههای β4 هضم نشده است.
در مواردی که میزان هموگلوبین S در هتروزیگوت داسی کمتر از ۳۵ درصد و یا هموگلوبین E کمتر از 21/5 درصد است بایستی در همراهی با فنوتیپ α0 یا هموزیگوت +α شک کرد. زمانی که هموگلوبین S در بیماری کمتر از ۲۱ درصد و E کمتر از ۱۵ یا ۲۰ درصد و میزان MCV بین ۵۰ تا ۵۵ است بایستی به همراهی ژنوتیپ Hبا هتروزیگوت داسی یا E فکر کرد، چون در این موارد هم ممکن است رسوب هموگلوبین اچ مشاهده نگردد.
کمبود سه ژن آلفا ایجاد بیماری هموگلوبین Hشبیه تالاسمی اینترمدیا میکند و کمبود چهار ژن آلفا با هیدروپس فتالیس و مرگ داخل رحمی همراهی دارد.
در کروماتوگرافی HPLC، هموگلوبینهای بارت و اچ ایجاد پیکهای اولیه میکنند و چنانچه نمونه خون آلوده به بیلیروبین باشد در همین منطقه پیک داده و با هموگلوبینهای بارت و اچ اشتباه میشود.
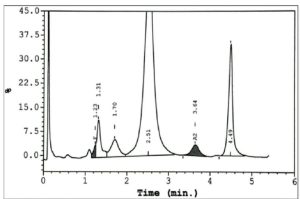
افزایش بیلیروبین موجب ایجاد باند در ناحیهی هموگلوبینهای بارت و H و هموگلوبین F استیله در کروماتوگرافی با روش HPLC میشود؛ از این رو انجام آزمایش با خون شستهشده با مشاهدهی باندهای یادشده توصیه میشود
فقدان 4 ژن آلفا که منجر به جنین هیدروپس میشود در حالتهایی که دو نفر با حذف سیس آلفا ازدواج کنند مشاهده میشود؛ دقت کنید که این حالت نیز از ازدواج فردی با بیماری هموگلوبین H با فردی با ژنوتایپ αα/– و نیز از ازدواج ژنوتایپ ααα/– با فرد α/αα- مشاهده شده است که در این حالت تشخیص قبل از ازدواج مشکل است زیرا ژنوتایپ ααα/– بهصورت فرد سالم ممکن است جلوه کند.
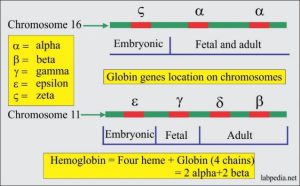
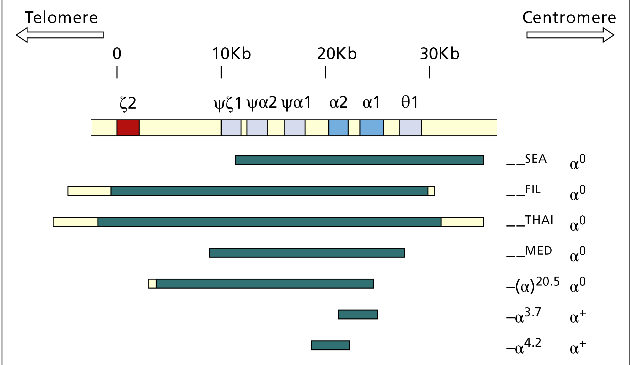
مهمترین ژنوتایپهای حذف سیس آلفا یا αα/– یا هتروزیگوت α0 مشاهده میگردد. خطوط آبی تیره منطقه حذفشده را نشان میدهند؛ برای مثال در حذف SEA و MED هر دو ژن آلفا حذف شده ولی ژن زتا حذف نشده ولی در هاپلوتایپ FIL علاوه بر حذف هر دو ژن آلفا، ژن زتا هم حذف شده است. مهمترین هاپلوتایپ تک آلفایی یا هاپلوتایپ +α با توجه به وسعت ناحیه حذف شده آلفا 3/7 و آلفا 4/2 است
از مهمترین ژنوتیپهای حذف سیس آلفا یا αα/– یا هتروزیگوت α0 میتوان به هاپلوتایــــپهای MED/αα ، —SEA/ αα- و –/ α20.5 اشاره کرد. ژنوتایپهای Fil/αα- و Thai/αα- با حذف ژنهای آلفا ۱ و آلفا ۲ و حذف ژن زتا روی هاپلوتایپ معیوب همراه است. جنین مبتلا به هموزیگوت Fil یا دوبل Fil و Thaiدر ماههای اول جنینی سقط میشود ولی چنانچه با نبود ژنهای آلفا، ژن زتا به ارث ببرد؛ برای مثال از ازدواج MED/αα ، —SEA/ αα– زندگی داخل رحمی تا تولد ممکن است ادامه یابد و نوزاد ورمکرده و هیدروپس اندکی پس از تولد فوت میکند.
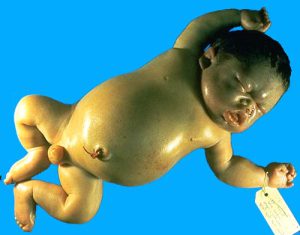
فقدان چهار ژن آلفا با زندگی ناسازگار است؛ زیرا سنتز هموگلوبینهای رویانی و جنینی و بزرگسالی به زنجیرهی آلفا وابسته است. در نبود زنجیرهی آلفا، هموگلوبینهای بارت (80 تا 90درصد) و مقداری هموگلوبین پورتلند و اندکی β4(HbH) در دوران جنینی تولید میشود. هموگلوبینهای یادشده به ترتیب H>Bart>Portland سریعتر از باند A روی استات سلولز در PH قلیایی حرکت میکنند. در هیدروپس فتالیس، گسترهی محیطی نوزاد مرده به دنیا آمده، انبوهی از گلبولهای قرمز هستهدار و میکروسیتوز را با تغییرات شدید شکل و اندازه نشان میدهد. هموگلوبین بارت به علت ناتوانی در پسدهی اکسیژن، جنین را با هیپوکسمی شدید مواجه میکند.
بیماری هموگلوبین H
بیماری هموگلوبین اچ با 4 سازوکار ژنتیکی بروز میکند:
- وراثت هتروزیگوت ترکیــــبی +α با ژنهای حذفی سیس آلفا مانند ژنوتایــــــــــــپهای α+/–SEA، -α4.2/–SEA، -α20.5/-α3.7 و MED/-α3.7– که در همه این موارد شخص فاقد سه ژن آلفاست.
- وراثت ترکیبی هاپلوتایپ α0 با آلفای غیر حذفی مثل آلفای کنستانت اسپرینـــــــــــگ α0/αCSα و یا MED/αTsaudiα–
- وراثت ترکیبی هاپلوتایپ α0 با ژن زنجیره بسیار ناپایدار آلفا مانند αQSα/–
- هتروزیگوت های ترکیبی غیر حذفی آلفا مانند αTsaudiα/ αTsaudiα و یا αTsaudiα/αAgrα
- جهش در ژن کنترلکننده سنتز زنجیره آلفا از راه دور که به نام ATRX نامیده میشود و محل آن روی کروموزوم X است.
کمبود زنجیره آلفا در دوران جنینی هر سه نوع هموگلوبینهای رویانی، جنینی و بزرگسالی را تحت تأثیر قرار میدهد. کمبود شدید آلفا در بیماری هموگلوبین H با تولد نوزاد کموزن همراه است. میزان MCV در ابتدای تولد بین 74-73 و MCH حدود ۲۲ است. الکتروفورز، الگوی F- BART- A را دارد و حدود ۲۰ تا ۴۰ درصد با میانگین ۲۰ درصد هموگلوبین بارت مشاهده میشود. بیشتر از 10 درصد هموگلوبین بارت در بدو تولد بیماری هموگلوبین اچ را مطرح میکند. پس از ۳ تا ۶ ماهگی هموگلوبین بارت ناپدید شده و هموگلوبین اچ با غلظت دو تا ۴۰ درصد (با میانگین ۸ تا ۱۰ درصد) جایگزین آن میگردد. میزان MCV و MCH و MCHC در بعد از دوران کودکی 65-50، 20-15 و 30-25 خواهد بود. میزان هموگلوبین بین ۸ تا ۱۰ گرم در دسیلیتر بوده و شدت کمخونی و طحال بزرگ در انواع غیر حذفی بیماری اچ شایعتر است.

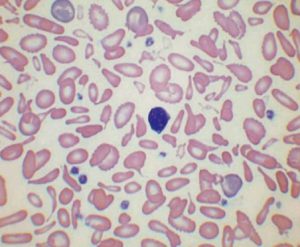
علائم بالینی بیماری هموگلوبین H متغیر است و گاهی آرام و گاهی وابسته به تزریق خون و گاهی با جنین هیدروپس همراه است. فقدان سه ژن آلفا، تصویری شبیه تالاسمی حد واسط یا اینترمدیا با گلبولهای قرمز میکروسیت و هایپوکروم، همراه با تغییرات شدید شکل و اندازه را به وجود میآورد. مقدار هموگلوبین حدود 3 گرم کمتر از افراد سالم از نظر گروه سنی است. الکتروفورز خون بند ناف یا نمونهی خون در دوران نوزادی بین یک تا 40 درصد، هموگلوبین بارت (γ4) را نشان میدهد و ممکن است بتوان مقدار اندکی هموگلوبین H را شناسایی کرد. در سندرمهای تالاسمی آلفا، محدود بودن زنجیرهی آلفا در دوران جنینی، با سنتز هموگلوبین بارت (γ4) و در دوران پس از تولد با هموگلوبین H (β4) همراه است.
گفتنی است بیشترین خطای افزایش کاذب پلاکت در بیماری هموگلوبین H رخ میدهد که به علت شمرده شدن میکروسیتوز شدید و گلبولهای شکسته بهجای پلاکت است. جالب آنکه با این خطای آزمایشگاهی میتوان هموگلوبین H را تشخیص داد؛ بدین معنا که هر وقت مرفولوژی میکروـهایپو و افزایش کاذب پلاکت، گاهی بهصورت ++++ یا حتی بالغ بر میلیون، توسط آنالیزور گزارش شد و تخمین پلاکت از روی گسترهی محیطی نرمال یا حتی کاهش داشت، آزمایش تهیهی اچبادی یا تهیهی مرفولوژی توپ گلف را انجام دهید یا باند سریع آن را در الکتروفورز مشاهده کنید.
هموگلوبینهای اچ و بارت فاقد تأثیر ارتباطheam-heam بوده و دارای میل ترکیبی زیاد برای اکسیژن هستند ولی اکسیژن پس نمیدهند. هموگلوبین اچ مستعد اکسید شدن در برابر داروهای اکسیدکننده است و شدت بیماری متعاقب مصرف اکسیدکنندهها، حاملگی و عفونت شدیدتر میشود.
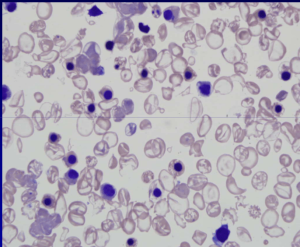
قبل از درآوردن طحال، رنگآمیزی حیاتی رسوب هموگلوبین اچ را بهصورت توپ گلف نشان میدهد ولی بعد از درآوردن طحال، هموگلوبین اچ بهصورت مجموعهای از اجسام هاینز بزرگ و توپ گلف مشاهده میشود. جسم هاینز H هم با رنگ رایت و هم با رنگ حیاتی رنگپذیر است.
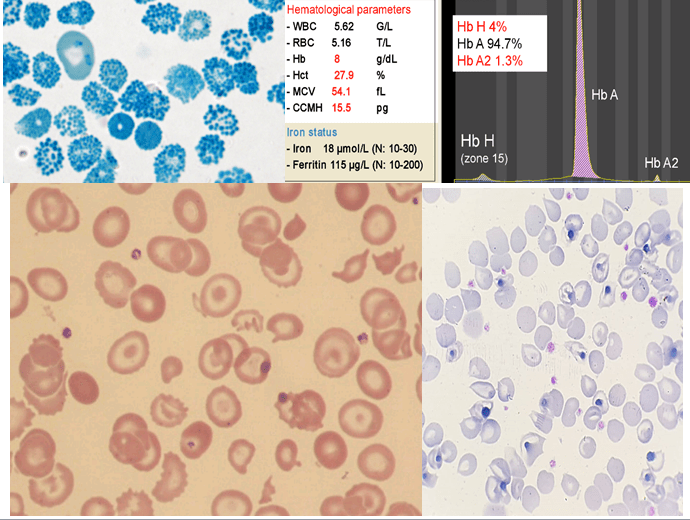
هموگلوبین H سریعترین باند را در الکتروفورز روی استات سلولز در PH قلیایی ایجاد میکند. برای تأیید آن میتوان نمونهی خون را با رنگآمیزی اکسیدکنندهی حیاتی، از قبیل کریزل بلوی درخشان انکوبه کرد و پس از نیم تا یک ساعت، آن را برای مرفولوژی توپ گلف یا اچبادی مشاهده نمود. اجسام H، رسوب دیناتورهی هموگلوبین H بوده که مانند فلفل پاشیده شده بر سطح گلبول نمایان میشوند. چنانچه رنگآمیزی، پس از برداشتن طحال صورت گیرد رسوب هموگلوبین H بهصورت اجسام هاینز بزرگ هم در رنگآمیزی حیاتی و هم در رنگآمیزی رایت دیده میشود. توجه داشته باشید که در بیماری هموگلوبین اچ مقدار MCV با میانگین 55 و MCH با میانگین 16 است.
همراه شدن ژنوتایپ هموگلوبین H با هتروزیگوت های داسی ، Cو E با تغییرات اندازه و اشکال متنوع گلبول همراه است. میزان MCV در این موارد کمتر از ۶۰ بوده و مقدار هموگلوبین غیرطبیعی به حدود ۲۰ تا ۲۵ درصد میرسد و در این موارد ممکن است حتی هموگلوبین اچ مشاهده نشود.
گفتنی است که زنجیرههای بتای سالم قابلیت تولید اچ و ایجاد تترامر β را دارند در حالیکه βE ایجاد تترامر نمیکنند و βs اضافی در سلول بهصورت رسوب درمیآید و در این موارد ممکن است تنها یک تا ۵ درصد گلبولهای قرمز رسوب اچ را نشان دهند.
سندرم ATRX با عقبافتادگی ذهنی، قد کوتاه و اختلال در ارگانهای جنینی همراه است و ممکن است که بهصورت هموگلوبین H بروز کند، گرچه ژنهایهای آلفا سالم هستند، ولی بیان نمیگردند.
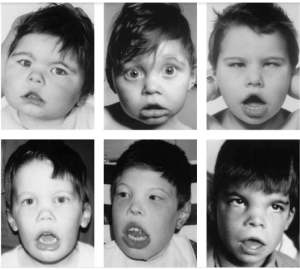
تالاسمی آلفا در دو سندرم متفاوت، با عقبماندگی ذهنی همراه است. در نوع اول، اختلال ذهنی خفیف و در برخی ناهنجاری اسکلتی مشاهده میشود. در این افراد در نتیجهی بازآرایی روی بازوی کوتاه کروموزوم 16 حذف وسیع (یک تا دو مگاباز) رخ داده است که هر دو ژن آلفا و همچنـین ژنهای کنار آن را حذف میکند. از این حالت با عنوان ATR-16 یاد میشود. AT به معنای آلفا تالاسمی و R مخفف retardation یا عقبافتادگی ذهنی است. جهش در ژن ATR-X روی کروموزوم Xq13.3 که فراوردهی آن هلیکاز است، موجب بروز کندذهنی و دیس مورفیسم و تالاسمی آلفا یا هموگلوبین اچ در افراد مذکر میشود. گمان میرود ژن ATR-X برای بیان ژنهای آلفا ضروری است.
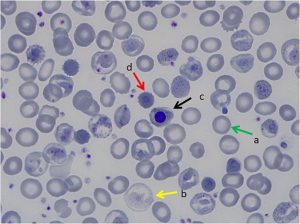
هموگلوبین اچ اکتسابی در سندرمهای مایلو دیسپلاستیک هم مشاهده میشود و ممکن است مقدار هموگلوبین اچ حتی تا ۶۰ درصد یعنی بیشتر از نوع ارثی مشاهده شود. در این موارد جهش در ژن ATRX نیز مطرح گردیده است.
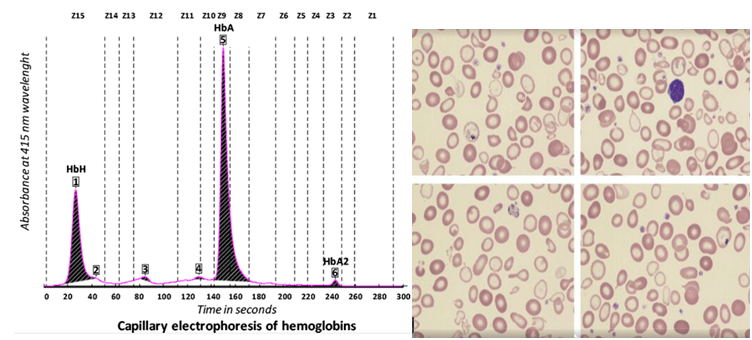
هموگلوبین اچ اکتسابی در سندرمهای مایلو دیسپلاستیک
به تصویر دای مورف خون محیطی بیمار توجه کنید
هموگلوبین کنستانت اسپرینگ:
جهش در رمز پایانی زنجیره آلفا موجب ترجمه شدن آن به اسید آمینه و طولانی شدن زنجیره آلفا با ۱۷۲ اسید آمینه میگردد. زنجیره آلفای نرمال دارای ۱۴۱ اسید آمینه است. سرعت سنتز کنستانت اسپرینگ بسیار کم است و در حالت هتروزیگوت حدود یک درصد هموگلوبین CS را تشکیل میدهد. ژن جهشیافته کنستانت اسپرینگ همیشه در کنار ژن سالم دیگر آلفا است.
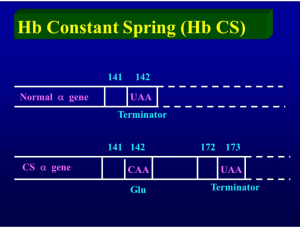
جهش در رمز پایانی ژن زنجیرهی آلفا، موجب ترجمهی بخشی از ناحیهی 3¢ژن تا رسیدن به رمز ختمکنندهی دیگری میشود. هموگلوبینهای کنستانت اسپیرینگ (TAA→CAA) و ایکاریا (TAA→AAA) و کویادورا (TAA→TCA) با 31 اسیدآمینهی اضافی در پایانهی زنجیرهی آلفا از نمونههای یاد شده است.
گرچه هتروزیگوت کنستانت اسپرینگ دارای علائم بالینی نیست ولی وراثــت هموزیگوت آن αCSα/αCSα ممکن است با بزرگ شدن طحال، ژاندیس و وابسته به تزریق خون باشد. در این موارد میانگین حجم گلبول قرمز نرمال است ولی کاهش MCHC به علت ورود آب به درون گلبول در نتیجه اکسیداسیون غشا دیده میشود.
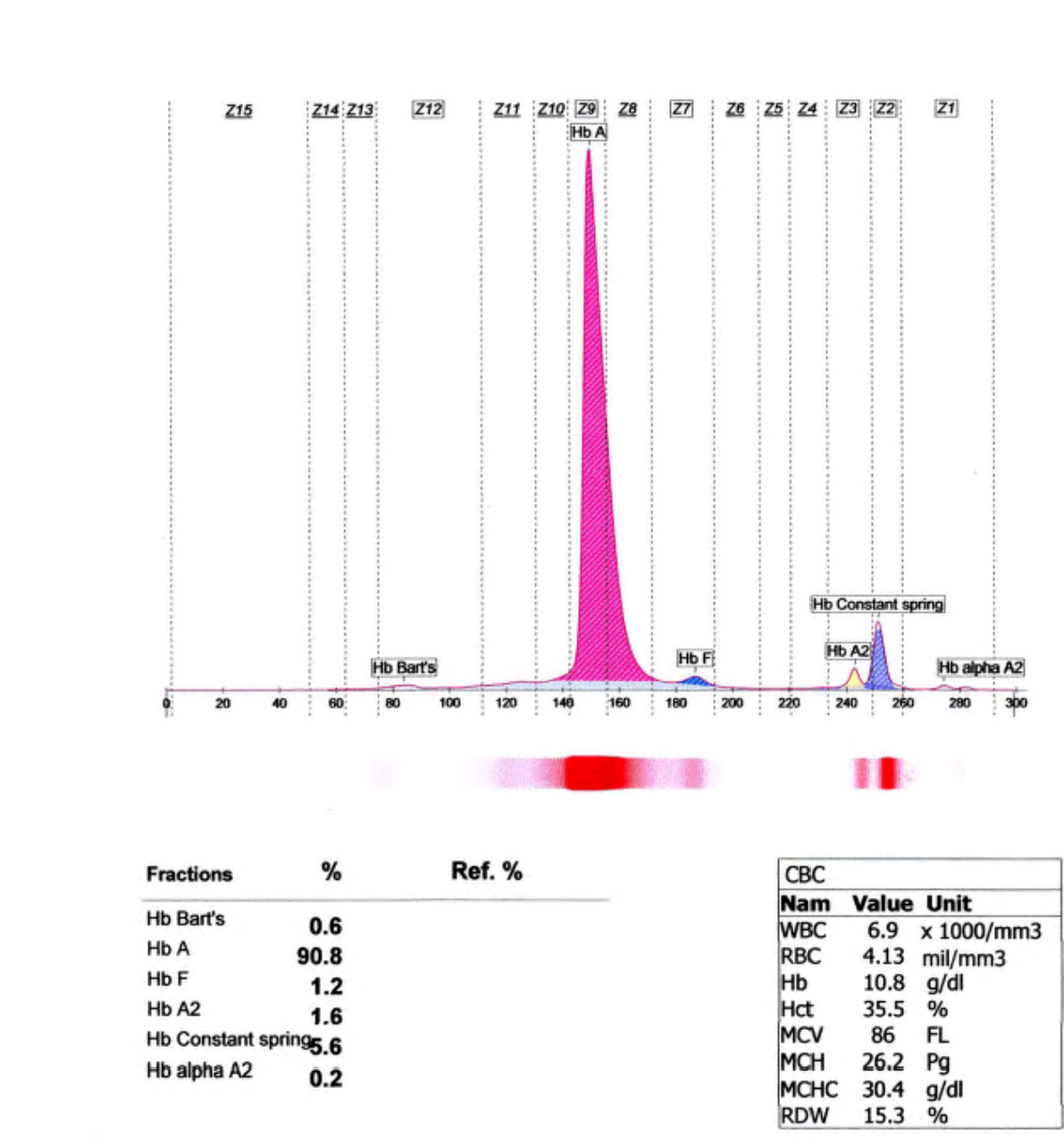
بیماران مبتلا به نوع هموزیگوت کنستانت اسپیرینگ (αcsα/αcsα) غالباً کمخون و با هموگلوبین حدود 10 گرم درصد، کاهش MCH حدود 26 پیکوگرم و مقدار نرمال یا نزدیک به نرمال MCV مواجهاند. گمان میرود که اکسیداسیون زنجیرههای غیرطبیعی آلفا موجب ورود آب به سلول و کاهش MCHC شود. افزایش تعداد رتیکولوسیتها، بین 6 تا 10 درصد و طحال و کبد بزرگ نیز مشاهده شده است. الکتروفورز هموگلوبین بین 2 تا 11 درصد هموگلوبین کنستانت اسپیرینگ (Hbcs)، بین یک تا سه درصد هموگلوبین بارت و بقیه از نوع A را نشان میدهد.
چنانچه شخص ژنوتیپ –/αCSα را به ارث ببرد، کمخونی شبیه تالاسمی اینترمدیا شدید با حضور هموگلوبینهای اچ و کنستانت اسپرینگ و بارت درمیآید. مواردی از این حالت ژنوتایپ با هیدروپس فتالیس با هموگلوبین بارت همراهی داشته است. هموگلوبین کنستانت اسپرینگ در الکتروفورز به روش سلولز استات در PH قلیایی بین باند کربنیک انیدراز و هموگلوبین A2 قرار میگیرد.
هموگلوبینهای کنستانت اسپرینگ و A’2 جزو هموگلوبین با حرکت کند بوده و هموگلوبین A’2 که ناشی از جهش در ژن دلتا است، بین کربنیک انیدراز و محل لکهگذاری روی استات سلولز قرار میگیرد. هموگلوبینهای کنستانت اسپرینگ و A’2 در الکتروفورز به روش کاپیلاری و HPLC جایگاه واضحتری دارند.
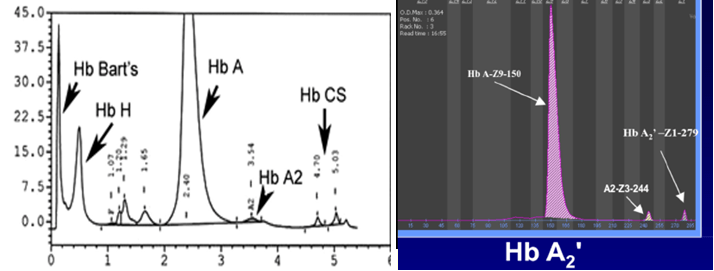
هموگلوبینهای کنستانت اسپرینگ و A’2 با کروماتوگرافی HPLC و روش کاپیلاری
هموگلوبینهای بارت و اچ در گروه هموگلوبینهای سریع هستند که بعد از A روی استات سلولز قرار میگیرند. برای جداسازی هموگلوبین H از بقیه هموگلوبینهای گروه Fast از رنگآمیزی حیاتی استفاده میشود. اکسید شدن هموگلوبین H بهصورت ذرات، سراسر گلبول را میپوشاند که به آن اچ بادی گویند. بعد از درآوردن طحال، اچ بادی بهصورت جسم هاینز بزرگی ظاهر میشود که هم با رنگهای رومانوفسکی و هم با رنگ حیاتی رنگپذیر میشود.
ژنو تایپ –/ααcs با هموگلوبین H و کنسانت اسپرینگ با کمخونی شدید همراه است. هموگلوبین کنستانت اسپرینگ کندتر از هموگلوبین A2 روی استات سلولز قرار گرفته ولی با کروماتوگرافی HPLC در دریچه C بهصورت پیک ریز قرار میگیرد. با تأخیر بیش از سه روز در انجام تست، شانس اینکه باندی مشاهده نشود وجود دارد.
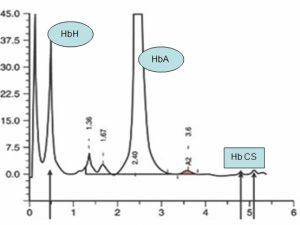
هموگلوبین H در همراهی با هموگلوبین کنستانت اسپیرینگ (–/αcsα) با کروماتوگرافی HPLC
هموگلوبین کنستانت اسپیرینگ با HPLC در دریچهی C جای میگیرد
از هموگلوبینهای سریع دیگر میتوان به هموگلوبین J اشاره کرد که یک واریان زنجیره بتا است و مواردی از آن در ایران هم گزارش شده است. هموگلوبین J یک واریان زنجیره بتا است و گفتنی است که جهش J طوری است که زنجیره آلفا میل بیشتری برای پیوند به آن نسبت به بتا دارد و از این رو بر خلاف واریانهای دیگر هموگلوبین، مقدار هموگلوبین J در حالت هتروزیگوت بیشتر از هموگلوبین A است.

موارد ضروری تشخیص قبل از تولد ابتلا به سندرمهای تالاسمی آلفا
واریانهای آلفای هموگلوبین:
در میان واریانهای آلفا که در کشور گزارش گردیده میتوان به هموگلوبینهای G، هموگلوبین آریا، هموگلوبین ستیف و هموگلوبینهای Q اشاره کرد.
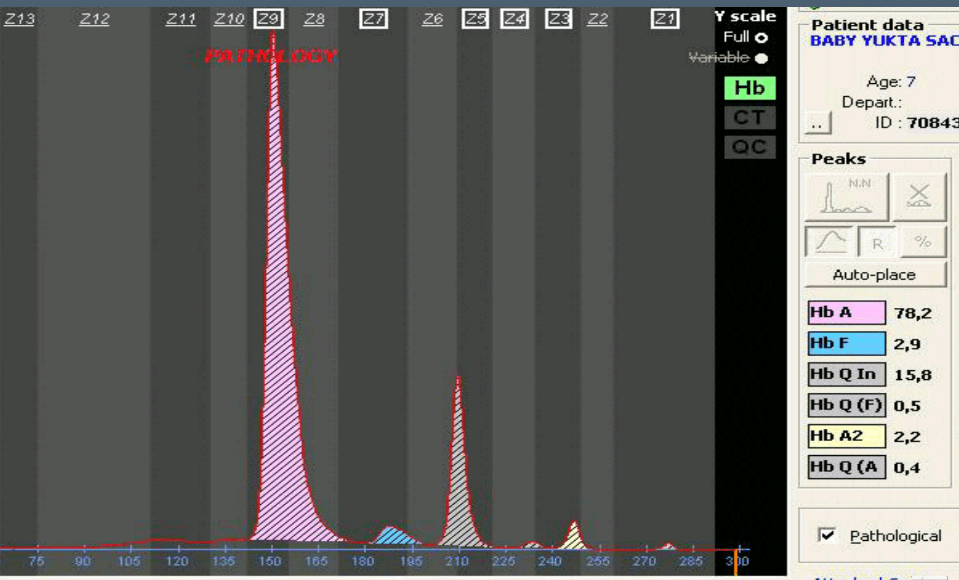
هموگلوبینهای Q و آریا و ستیف در الکتروفورز با استات سلولز روی باند S قرار میگیرند و مقدار آنها در حالت هتروزیگوت کمتر از ۲۵ درصد است، با الکتروفورز با روشهای کاپیلاری و HPLC میتوان باندهای ماینور و کوچک دو بخشی A2 + G2 و یا A2+ arya2 و یا A2 + Q2 را مشاهده کرد.
غلظت واریانهای آلفا بستگی به تعداد جهشهای ژن آلفا دارد؛ برای مثال فنوتیپ αG/ -αG – با بیش از 95 درصد هموگلوبین G شبیه تالاسمی ماینور آلفا بروز میکند. الکتروفورز باند G2 هم که ارزشی معادل A2 دارد نشان میدهد.
واریان هموگلوبینهای آلفا غالباً از نظر بالینی آرام هستند. هموگلوبین ستیف (Setif) در خارج از بدن در محلول بافری رسوب و ایجاد گلبولهای شبهداسی میکند که به نام سیکل کاذب (Pseudosickling) مشهور است.
واریانتهای هموگلوبین و اهمیت بالینی آنها
تاکنون بیش از 1150 واریانهای مختلف زنجیره آلفا و بتا ناشی از جهشهای گوناگون شناخته شده است که برخی آرام و برخی حالت تهاجمی دارند. گرچه شناسایی جهشهای آرام از نظر بالینی بدون اهمیت است ولی گاهی این هموگلوبینها ایجاد پیک یا باند و یا همپوشی در جایگاه الکتروفورز با هموگلوبینهای تهاجمی دارند و از این رو افتراق آنها لازم است.
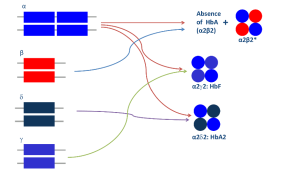
با توجه به اینکه زنجیره آلفای گلوبین با زنجیرهای بتا و دلتا و گاما ایجاد هموگلوبینهای A و A2 و F میکند از این رو یک واریان آلفا میتواند سه نوع واریان هموگلوبین علاوه بر هموگلوبینهای نرمال A و A2 و F را ایجاد کند. زنجیره بتا تنها با زنجیره آلفا پیوند میخورد و از این رو یک واریان ژن بتا ایجاد یک واریان هموگلوبین و هموگلوبین نرمال A میکند. واریانهای دلتا و گاما هم مشاهده شده است. واریانهای هموگلوبین A2 هم ناشی از جهشهای ژن دلتا و هم ناشی از جهشهای ژن آلفا تولید میشوند و شناسایی آنها اهمیت فراوانی برای شناخت تالاسمی ماینور بتا و شناخت واریانهای آلفا دارد.
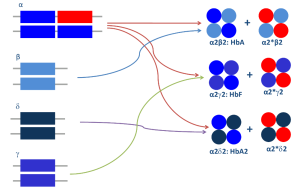
با توجه به اینکه زنجیره آلفای گلوبین با زنجیرهای بتا و دلتا و گاما ایجاد هموگلوبینهای A و A2 و F میکند از این رو یک واریان آلفا میتواند سه نوع واریان هموگلوبین علاوه بر هموگلوبینهای نرمال A و A2 و F ایجاد نماید.
برای مثال واریان دلتا ایجاد هموگلوبین A’2 و واریان آلفا مثل αG با زنجیره دلتا ایجاد هموگلوبین G2 میکند. در این موارد در الکتروفورز هموگلوبین باند دوبخشی A2 (Split A2) ایجاد میکند؛ بدین مفهوم که جایگاه A2 نرمال از جایگاه A’2 و G2 متمایز میگردد و در حالی که کل هموگلوبین A2 برابر با A2 + A’2 و یا بـــــــرابر G2 +A2 است، چنانچه این حالت مورد شناسایی قرار نگیرد مقدار کل هموگلوبین A2 را کمتر گزارش کرده و تشخیص تالاسمی ماینور بتا با مشکل مواجه میشود.
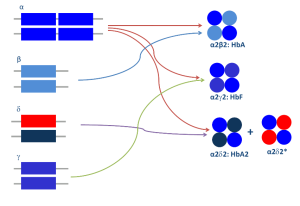
واریانهای هموگلوبین A2 هم از جهشهای ژن دلتا و هم از جهشهای ژن آلفا تولید میشوند
در حضور یک واریان زنجیره بتا برای مثال واریان داسی(βS) انتظار میرود میزان هموگلوبینهای A و S با هم برابر و حدود ۵۰ درصد باشد ولی توجه داشته باشد که زنجیره آلفای سالم به علت شارژ مثبت کشش بیشتر برای پیوند با زنجیره بتای سالم که الکترونگاتیو است را دارد. در مواردی از قبیل واریانهای βS و βC که جهش موجب افزایش بار مثبت زنجیره شده است به علت اینکه زنجیره آلفا تمایل بیشتری برای بتای سالم دارد مقدار هموگلوبین واریانت کمتر از ۵۰ درصد میگردد و بین ۴۰ تا 45 درصد میشود، در این موارد چنانچه هموگلوبین واریان بیشتر از ۴۵ درصد تا حدود ۵۰ درصد قرار گیرد ممکن است با هاپلوتیپهای سهتایی ژن آلفا مواجه باشیم.
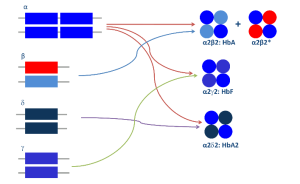
جهش در یک ژن بتا تنها تولید یک واریان هموگلوبین میکند که در صورت ژنهای سالم آلفا مقدار آن بین 40 تا 45 در صد است که برای مثال میتوان به هتروزیگوت داسی اشاره کرد
مقدار واریان هموگلوبینهای زنجیره بتا بستگی به تعداد ژنهای آلفا هم دارد، برای مثال در حضور ۴ ژن آلفا مقدار هموگلوبین S در هتروزیگوت داسی ۴۰ درصد و در حضور سه یا دو و یک ژن آلفا مقدار هموگلوبین S به ترتیب به 35 و ۲۸ و ۲۰ درصد افت میکند.
جهش یکسان در ژنهای بتا تولید یک واریان غیرطبیعی هموگلوبین میکند که برای مثال میتوان به هموزیگوت داسی اشاره کرد
سرعت سنتز زنجیره نیز از موارد تعیینکننده واریان هموگلوبین است؛ برای مثال واریان βE با سرعت کندتری ساخته میشود و مقدار آن در حالت هتروزیگوت با حضور ۴ ژن آلفا برابر ۲۵ تا ۳۵ درصد است. واریان لپور با سرعت سنتز کم در حالت هتروزیگوت ۱۰ تا ۱۵ درصد کل هموگلوبین را تشکیل میدهد.
به علت میل ترکیبی بیشتر زنجیرهی آلفا برای زنجیرهی سالم β، مقدار هموگلوبین A در هتروزیگوت داسی بیشتر از 50 درصد است و مقدار هموگلوبین S حدود 40 درصد است. مقدار HbS، متأثر از تعداد ژنهای آلفاست. هتروزیگوت داسی در کنار دو ژن آلفا مقدار هموگلوبین S را به 35 درصد و در همراهی هموگلوبین (–/α-) H مقدار HbS را به حدود 10 تا 25 درصد میرساند. در همراهی هاپلوتایپهای αα/ααα با هتروزیگوت داسی مقدار HbS حدود 45 تا 50 درصد میشود. چنانچه جهشهای تالاسمی و داسی روی یک ژن بتا در حالت هتروزیگوت قرار گیرند، ممکن است مقدار HbS کمتر از 10 درصد شود. موارد فوق در مورد هموگلوبینهای C و E هم صادق است. چنانچه مقدار هموگلوبین غیرطبیعی بیشتر از هموگلوبین A شود و گلبولهای قرمز میکروسیت و هایپو کروم باشند. همراهی واریانت هموگلوبین با ژن تالاسمی مطرح میگردد.
گفتنی است که واریان هایβJ مانند J Iran و J Batimore از نظر شارژ الکترونگاتیو هستند و تمایل بیشتری برای پیوند به آلفا دارند و از این رو مقدار هموگلوبین J بیشتر از ۵۰ درصد کل هموگلوبین میشود.
پیشبینی مقدار واریان آلفا پیچیدهتر از بتا است. هر شخص سالم دارای ۴ ژن آلفا است. دو ژن α2 و دو ژن α1 ژنوتایپ آلفای شخص سالم را شکل میدهند. ژنهای α2 حدود ۷۰ درصد و ژنهای α1 حدود ۳۰ درصد فراورده زنجیره آلفا را به عهده دارند، از این رو یک واریانت آلفا ممکن است یک واریان 12/5 درصدی تا 37/5 درصد از کل هموگلوبین را تشکیل دهد.
در هتروزیگوت داسی، تنها یک ژن بتا معیوب است که میتوان آن را بهصورت βS/βA نمایش داد. وراثت داسی هتروزیگوت، معمولاً بدون علائم بالینی و هماتولوژی است؛ بههرحال پدیدهی داسی شدن ممکن است در مواقعی از قبیل تب بالا، هیپوکسمی بسیار شدید، کوهنوردی، ورزشهای سنگین و بیهوشی رخ دهد. مواردی از مرگ ناگهانی در رابطه با ورزشهای سنگین در همراهی با کمآب شدن شدید بدن و اسیدوز گزارش شده است.
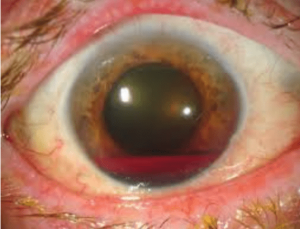
خونریزی در قسمت قدامی چشم بر اثر تروما یا بیماریهای چشم را هایفیما گویند. چنانچه این آسیب در هتروزیگوت و هموزیگوت داسی رخ دهد به علت اسیدی بودن این محل، پدیده داسی شدن رخ داده و فشار داخل افزایش مییابد که ممکن است آسیبهای غیر قابل برگشت را در پی داشته باشد و نیاز به مداخله جراحی باشد. از این رو آزمایش داسی برای بیماران با هایفیما سفارش میشود.
تشکیل خودبهخودی گلبولهای داسی در هتروزیگوت داسی در پاپیلای کلیه ممکن است موجب نکروز پاپیلاری و هماچوری و کاهش قدرت تغلیظ ادرار و شب ادراری شود. همراه شدن تالاسمی آلفا با هتروزیگوت داسی، به علت کم کردن تراکم هموگلوبین S در گلبولها، موجب بهتر شدن قدرت تغلیظ میشود.
افزایش شیوع باکتری اوری و پیلونفریت و سندرمهای افزایش فشار خون در زنان باردار با هتروزیگوت داسی مشاهده شده است. گفتنی است ارتباطی چشمگیر بین کارسینوم مدولاری کلیه و هتروزیگوت داسی وجود دارد.
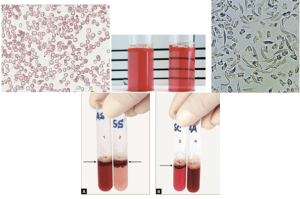
هرچند گلبولهای داسی در هتروزیگوت داسی در خون مشاهده نمیشود، ممکن است تعدادی گلبولهای قرمز فربه که دو طرف تیز دارند، مشاهده شود. اینگونه سلولها (plump RBC) در 96 درصد موارد هتروزیگوت داسی هستند و در 4 درصد اشخاص سالم گزارش شده است. آزمایش حلالیت برای دستیابی به نتیجهی مثبت، به حداقل 20 درصد هموگلوبین S نیاز دارد. افزودن گلبولهای قرمز حاوی هموگلوبین داسی به بافر فسفاته ساپونیندار با مولاریته 2/3 موجب کدرشدن و مانع از مشاهدهی خطوط کارت در پشت لوله میشود. لکوسیتوز، افزایش پاراپروتئین، لیپیدمی، اجسام هاینز، هموگلوبینهای بارت و ناپایدار و هموگلوبین ستیف و I موجب مثبت کاذب در آزمایش میشود. افزودن گلبول قرمز شستهشده، بسیاری از خطاها را کاهش میدهد.
هموزیگوت داسی (βSβS) با الگوی الکتروفورز S+F با مقدار متغیر HbA2 همراه است. مقدار هموگلوبین F غالباً بین 1 تا 20 درصد متغیر است. پروسهی داسی شدن گلبولهای قرمز در فشار کم اکسیژن، چند بار برگشتپذیر است؛ ولی با تثبیت آسیب به غشا، بهصورت داسی برگشتناپذیر درمیآید.
در سلولهای داسی برگشتناپذیر، کلسیم افزایش و پتاسیم و آب کاهش مییابد و سیالیت سلول از بین میرود. آسیبهای اکسیداسیون به غشای گلبولهای قرمز و پلیمری شدن هموگلوبین موجب خوشهای شدن باند 3 و اتصال IgG و فاگوسیتوز آنها میشود. افزایش بیان مولکولهای چسبندگی روی سلولهای داسی موجب چسبیدن آنها به سلولهای اندوتلیال و بحرانهای انسداد عروقی میگردد.
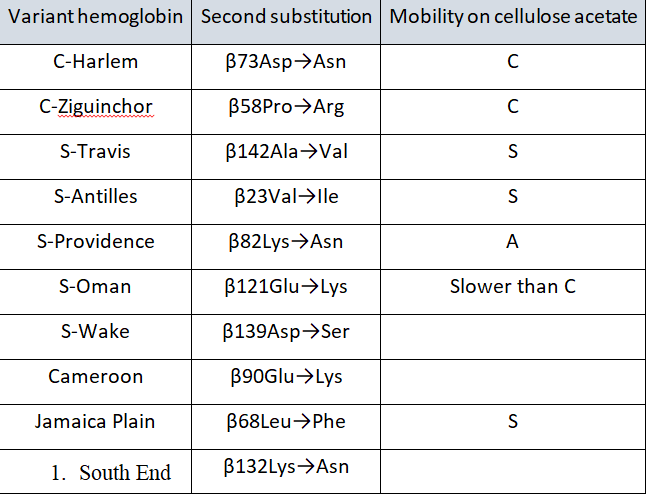
گفتنی است سه واریان هموگلوبین با دو جهش در زنجیرهی بتا که جهش داسی (β6glu®Val) یکی از آنهاست، قادر به داسی شدن در حالت هتروزیگوت است. این سه واریان عبارتاند از: هموگلوبین S آنتیل و هموگلوبین S عمان و جامائیکا
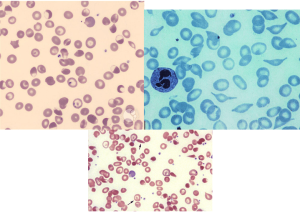
سلول داسی به اشکال گوناگون، از قبیل داسی کلاسیک، داسی فربه، کلاه ناپلئونی، دوکیشکل با انتهای نوکدار در دو طرف و قایقی شکل و تاولدار، مانند بایتسل کشیدهشده مشاهده میشوند. اشکال کلاه ناپلئونی در داسی عمان مشاهده میشود.
هتروزیگوتهای ترکیبی داسی و تالاسمی (β0βS و β+βS)
وراثت همزمان یک ژن تالاسمی β و یک ژن داسی موجب شکلگیری هتروزیگوت ترکیبی داسی و تالاسمی میشود. ویژگی این حالت ترکیبی به قرار زیر است:
- شدت کمخونی کمتر از آنمی داسی شکل است.
- کاهش اندکسهای (MCH, MCV) RBC؛ برخلاف هموزیگوت داسی که با مرفولوژی نرموسیت و نرموکروم همراه است.
- حضور تارگتسل در گسترهی محیطی چشمگیرتر از هموزیگوت داسی است. بیشترین مقدار تارگتسل تا 90 درصد، در هموزیگوت هموگلوبین C و حدود 30 درصد در هموزیگوت داسی دیده میشود. گلبولهای داسی هیپوکروم، بهویژه از نوع دوکی و قایقی در گسترهی محیطی دیده میشوند.
- طحال بزرگ در حالت +βSβ ممکن است تا بزرگسالی باقی بماند و بیمار مستعد آنفارکتوس طحال در شرایط هیپوکسی شود.
الکتروفورز در حالت +βSβ بیشتر از 50 درصد هموگلوبین S را نشان میدهد و همیشه برخلاف هتروزیگوت داسی مقدار HbS بیشتر از HbA است. مقدار هموگلوبین A از 5 تا 30 درصد و HbF از 5 تا 15 درصد و HbA2 از 3/5 تا 5/5 درصد متغیر است.
الکتروفورز در حالت βSβ0 الگوی SF دارد و کاهش اندکسها نیز شدیدتر از +βSβ است. در این حالت، هموگلوبین A مشاهده نمیشود.
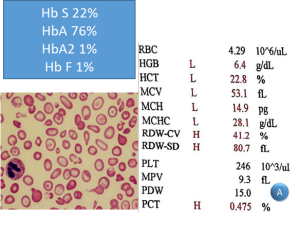
توجه داشته باشید هنگامی که ژنوتایپ اچ (فقدان سه ژن آلفا) با هموگلوبینوپاتیهای دیگر از قبیل هتروزیگوت داسی و یا D و یا O عرب و یا E به صورت همزمان به ارث برسند، ممکن است که رسوب اچ بادی با رنگهای حیاتی دیده نشود. در این موارد از روی غلظت هموگلوبین غیرطبیعی برای مثال هموگلوبین S حدود 20 درصد و اندکسهای بسیار پایین، پِی به همراهی ژنوتایپ اچ برده میشود و نیاز به مطالعه مولکولی را مطرح میکند. در بیمار فوق کمخونی ترکیب هموگلوبین اچ و هتروزیگوت داسی دیده میشود.
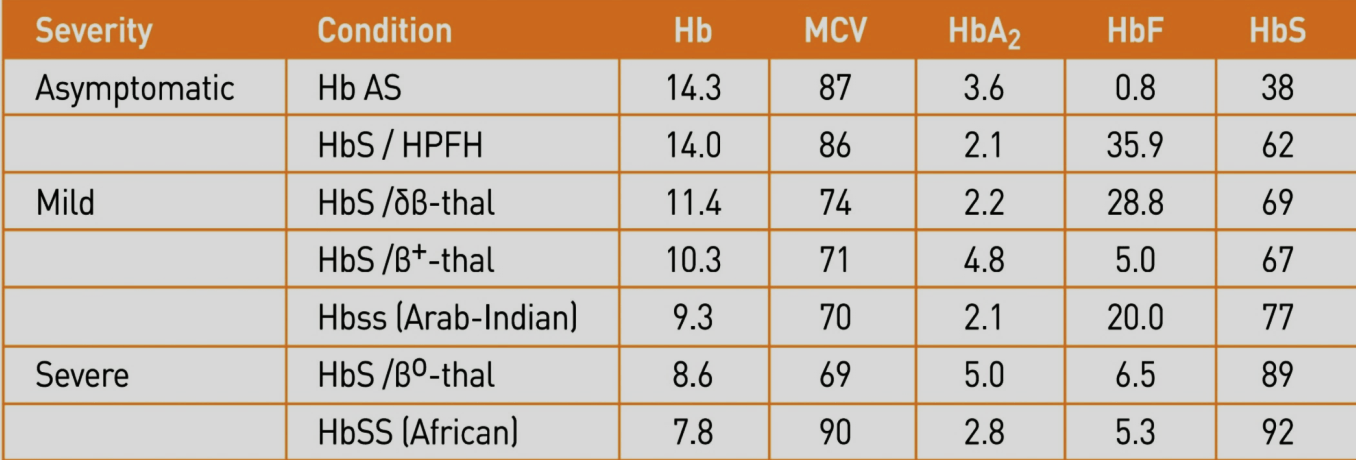
پارامترهای خون در هتروزیگوتهای دوبل داسی و تالاسمی را مشاهده میکنید. به غلظت هموگلوبین F در موارد مختلف توجه کنید. گفتنی است که هموگلوبین F بیشتر از 30 درصد آنمی داسی را آرام و حتی بدون علامت میکند.
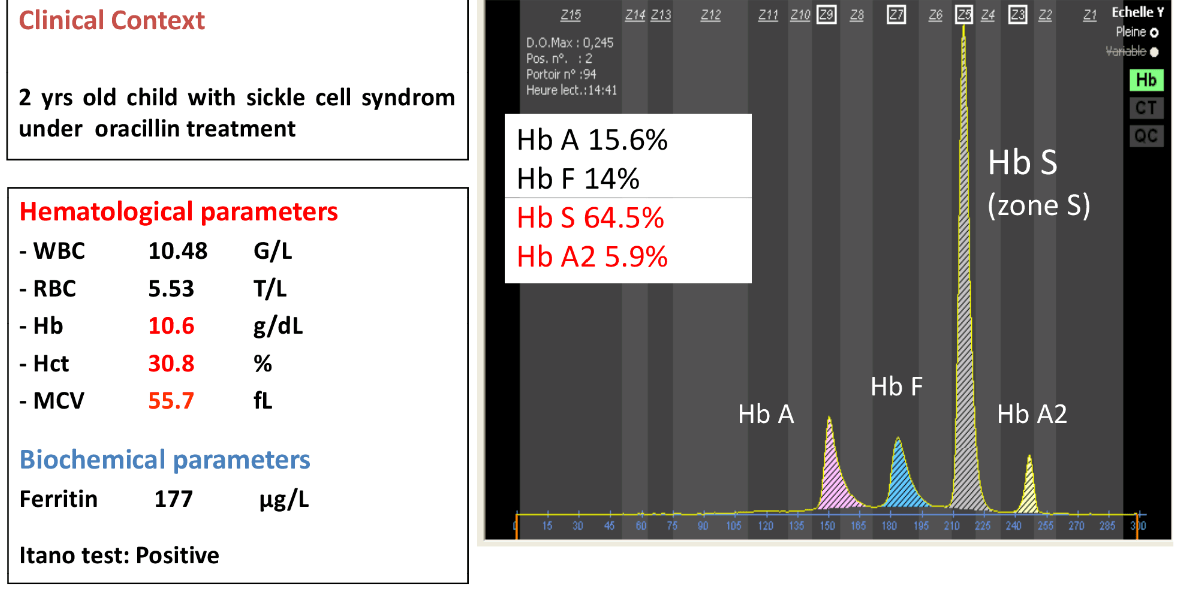
الکتروفورز در حالت +βSβبیشتر از 50 درصد هموگلوبین S را نشان میدهد و همیشه برخلاف هتروزیگوت داسی مقدار HbS بیشتر از HbA است. مقدار هموگلوبین A از 5 تا 30 درصد و HbF از 5 تا 15 درصد و HbA2 از 3/5 تا 5/5 درصد متغیر است.
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام