رمضان اکبریان _ مهدی حقدوست – استاد راهنما جناب دکتر امیر سید علی مهبد
چکیده:
هموگلوبینوپاتی یکی از مهمترین بیماریهای ارثی است که مواجهه با آن نیاز به داشتن آمار درست مبتلایان به آن و شناخت نوع هموگلوبینوپاتی آنها دارد. خوشبختانه از حدود 20 سال قبل آزمایشهای مناسب برای غربالگری این بیماری و جلوگیری از انتقال ارثی آن در ایران انجام میشود. در این مقاله پژوهشی سعی شده است که شیوع این بیماری در شهر بندرعباس و در کنار آن کارایی روشهای غربالگری بررسی شود و این مطالعه به روش توصیفی تحلیلی و مقطع زمانی یکساله از آذر 1392 تا آذر 1393 انجام گرفته است. بیماران افراد تحت بررسی بودند که به بیمارستان خاتمالانبیاء مراجعه نموده و درخواست بررسی هموگلوبینوپاتی داشتهاند و طبق برنامه غربالگری کشوری، بیماران باردار و بیماران دچار فقرآهن از مطالعه حذف گشته و بقیه افراد تحت بررسی با الکترفورز قلیایی با PH=8.6 و در صورت نیاز با تستهای تأییدی برای HbA2 وHbF و HbS بررسی شدند. نتایج بعضی از بیماران بر حسب نیاز با روش (Capillary Electrophoresis) نیز کنترل گشته است. نمونهها شامل 5838 نفر بودند که 85 خانم باردار و 290 نفر بعلت فقرآهن از مطالعه حذف شدند. شیوع کمخونی ارثی در بندرعباس 9.4% به دست آمد که شامل 33.3% HbA2>3.5، 3.4% HBF،7.5 %HbS،11.1 %HbD و یک مورد HbH با 0.2% و یک مورد هموگلوبین Constant Spring با 0.2% و حدود 44.3% هموگلوبینوپاتی ناشی از زنجیره آلفا یا ناشناخته بوده است. با توجه به شیوع یکسان انواع هموگلوبینوپاتی در سنین 20-1 سال، 50-20 سال و 50 سال به بالا حداقل ثابت میشود که غربالگری هموگلوبینوپاتی در موقع ازدواج در شیوع هموگلوبینوپاتی بیتأثیر بوده است.
با احتمال بیشتر از 95% نشان داده شده است که افزایش HbA2 با افزایش HbD وHbF همراه بوده است، یعنی هر چه فراوانی HbD وHbF بیشتر میشود احتمال وجود و همراهی افزایش HbA2 نیز بیشتر میشود، پس بیشتر موارد هموگلوبینوپاتیهایD و F با ژن تالاسمی بتا نیز همراه هستند.
واژگان کلیدی: هموگلوبینوپاتی – بندرعباس
مقدمه:
طبق تعریف دیویدسون (دیویدسون 2007) اگر غلظت هموگلوبین یا هماتوکریت کمتر از پایینترین حد 95 درصد محدوده مرجع (با توجه به سن- جنس- موقعیت جغرافیایی{ارتفاع}) برسد کمخونی تلقی میشود. البته کمخونی ممکن است مطلق یا نسبی باشد یا پاتولوژیک یا فیزیولوژیک بوده و یا ممکن است علامتی از یک بیماری زمینهای باشد.
کمخونی پاتولوژیک به کمخونی فقرآهن، مگالوبلاستیک، کمخونی بیماری مزمن، بیماری کلیوی کبدی و اندوکرینی، میلوفیتزیک، آپلاستیک، ایدیوپاتیک، سیدروبلاستیک، متعاقب خونریزیهای حاد و انواع همولیزها و هموگلوبینوپاتیها تقسیمبندی میشود. بیش از 75 درصد این هموگلوبینوپاتیها به علت جایگزینی یک اسید امینه در زنجیره آلفا و یا بتای زنجیره گلوبین رخ میدهد که علت آن جایگزینی یک باز خاص در کدون سه حرفی ژن مربوطه است که بعلت termination error (خطای اختتام)،Fram shift جهش انحــــرافی قالبی، کراسینگاور cross over in phas حذف کدون یا زنجیره آمیخته شده fused یا هیبرید شده زنجیره پلیپپتیدی بطور ناهنجاری بلند یا کوتاه است. در بعضی مواقع نیز جایگزینی یک باز باعث نتایجی در نسخهبرداری آن میشود که شامل کاهش تولید mRNA یا شکلگیری mRNA ناپایدار میشود، ماحصل این روند تولید زنجیرههای آلفا یا بتا ناپایدار است. در مطالعات جدیدتر هموگلوبینوپاتی را از طریق وجود MiRNA در نمونههای بیماران شناسایی مینمایند که تمایز و القا به یک رده خاص را افزایش میدهند (سید امیرحسین امامی 1390)
کمخونی از جمله شایعترین مشکلات ارثی و تغذیهای در جهان محسوب میشود (اصنافی 1382) که بیش از 2 میلیارد نفر از مردم جهان را گرفتار ساخته است. (مهدی حقی 1388) به غیر از موارد ارثی، کمخونی رابطه مستقیمی با وضعیت اجتماعی، اقتصادی، منطقه جغرافیایی و شغلی افراد دارد؛ کمخونی باعث تغییر در روند زندگی افراد و در موارد شغلی موجب تغییر قابلیتهای جسمی و ذهنی و اختلال در توانایی افراد و علائم بالینی مانند سرگیجه، خستگی زودرس و در موارد شدید باعث آنژین صدری میشود. (الهام ایمانی 1392)
در میان کمخونیها1 انواع ارثی جایگاه ویژهای دارند. با توجه به تنوع هموگلوبینوپاتیها شناسایی نوع ارثی و دستهبندی آن اهمیت ویژهای دارد. تشخیص نوع کمخونی و علت آن و افتراق انواع اکتسابی از موارد ارثی از اهمیت بالایی برخوردار است. بیش از 800 هموگلوبین جهش یافته انسانی وجود دارد. جهشها باعث ناپایداری در ساختمان هموگلوبین، افزایش یا کاهش میل اتصال آن با اکسیژن یا افزایش در سرعت اکسیداسیون آهن دو ظرفیتی (Fe2+) هِم به وضعیت فریک (Fe3+) (بیوشیمی دولین 2011)، یا تغییر در بار الکتریکی هموگلوبین جهش یافته و تغییر در الگوی حرکت الکترفورزی آن میشوند.
ناقلین بیماری تالاسمی ممکن است از نظر آزمایشگاهی خصوصیات فنوتایپی مختلفی داشته باشند (طیبه چهکندی 1381)، شدت هموگلوبینوپاتی از وضعیت بدون علامت و فقط دارای اختلالات آزمایشگاهی تا بروز مرگ در رحم مادر متغیر است. برخلاف تالاسمی آلفا که علت ان معمولاً حذفی است بتا تالاسمی عمدتاً ناشی از جهش نقطهای (Point Mutaion) میباشد. خصیصه بتا تالاسمی میتواند به علت حذف ژن و یا تولید کم فراوردههای آن رخ دهد گرچه این افراد از نظر بالینی سالم هستند ولی از نظر آزمایشگاهی شاهد کاهش در مقادیر MCV و MCH آنها هستیم. در حالت هموزیگوت، بتا تالاسمی میتواند از یک کمخونی خفیف (کاهش در تولید دو ژن بتا) تا تالاسمی متوسط (به ارث رسیدن یک ژن بتای آفریقایی، فقدان یک ژن بتا و یا به ارث رسیدن دو ژن بتای مدیترانهای) و حالت شدید وابسته به انتقال خون (فقدان دو ژن بتا) متغیر باشد یا زنجیره آلفای مازاد در پیشسازهای اریتروییدی رسوب نموده سبب خونسازی غیرمؤثر میشود. ژنتیک آلفا تالاسمی پیچیده بوده و علت نقص ژنتیکی در تمام نژادها یکسان نیست. اغلب جهشهای ایجاد شده حذفی بوده و در پارهای از موارد جهشهای غیرحذفی نیز گزارش شدهاند. (علی موحد 1387) پیشنهاد شده است با توجه به تنوع قومی و تعداد نسبتاً زیاد جهشهای ژنی، انجام بررسی بیشتر در همه نقاط ایران برای ردیابی ناقلین و بسته به روش اندازهگیری، حساسیت دستگاهها و خصوصیات ژنتیکی هر جامعهای الزامی شود. تعیین معیارهای تشخیصی جهت شناسایی ناقلین هر یک از جوامع ضروری است.
تشخیص قطعی انواع تالاسمی نیاز به آنالیز دقیق DNA دارد ولی به دلیل محدودیت امکانات در برنامه غربالگری کشوری تالاسمی از معیارهای MCV<80fl و MCH<27pg و تعیین HbA2 استفاده میشود. در صورتی که فردی مشکوک به بتا تالاسمی مینور باشد، اندازهگیری HbA2 ضروری است (منیره عطاپور 1382). بودن MCV بالاتر از 80 در بتا تالاسمی مینور معمول نبوده ولی در صورت اندازهگیری میزان 80 MCV< حتی در صورت طبیعی بودن دیگر شاخصهای اریتروسایتی و HBA2 احتمال تالاسمی Intermedia وجود دارد. در این افراد به احتمال زیاد یکی از ژنهای آلفا تالاسمی وجود دارد که با آزمایشهای معمول غیرقابل تشخیص بوده و به نام Silent Carrier نامیده میشوند. (منیره عطاپور 1382)
حتی در بیماری هموگلوبین (H) با وجود MCV<78 و MCH<27، ممکن است شاهد الکتروفورز طبیعی باشیم. در این موارد عوارض بالینی کمخونی همولیتیک و هپاتواسپلنومگالی میتوانند در تشخیص بیماری کمک کننده باشند. (خدامراد زندیان 1380) طبق آخرین پژوهشهای انجام گرفته در بین نمونه افرادی که به طور تصادفی انتخاب شدند، افراد هتروزیگوت که دارای هموگلوبینوپاتیهایی بوده ولی دارای درصد HbA2 در حد طبیعی بودهاند 72.4% برآورد شده است. (ابراهیم میری مقدم 1391)
در یکی از پژوهشها ثابت شده است که خونریزیهای حاد و یا بعد از اعمال جراحی سنگین نیز میتواند باعث کاهش هموگلوبین شود. (جعفر نصوحی 1382)
در این بررسی بعلت پیچیدگی بررسی آنمی در زنان و رابطه غیرمتعارف شرایط بالینی مادران و نوزادان و در عین حال افزایش مقادیر مطلق هموگلوبین در دوران بارداری و نوزادی (زهرا حسینی بهارانچی 1383) مادران باردار از مطالعه حذف شدهاند.
ایران بر روی کمربند جغرافیایی تالاسمی قرار دارد. میزان ناقلین ژن تالاسمی را در جزایر ساردین 11-44٪، سیسیل 15٪، یونان 5 تا 15٪، ایتالیا 4 تا 10٪ و ایران 4 تا 10٪ برآورد نمودهاند. (زندیان1388) حدود 2 میلیون نفر در کشور حامل ژن بتا تالاسمی هستند. نواحی دریای خزر و خلیج فارس با 10٪ بیشترین فراوانی را به خود اختصاص دادهاند. برطبق بررسیهای انجام شده ازدواج فامیلی در استان هرمزگان حدود 38٪ میباشد، 27٪ آنها در میان عموزادهها و عمهزادهها که زادههای نخست هستند میباشد. (حسینپور فیضی.1388) نسبت بالای ازدواج فامیلی در این استان (هرمزگان) و پیامد آن در خزانه ژنی موجب بروز اشکال شدید و پیچیده و یا بسیار خفیف و بدون علایم شده است. (حسینپور فیضی.1388) در بررسی هموگلوبینوپاتیها عمل بعضی از داروها که تأثیر بسزایی در ترکیب هموگلوبین دارند را باید مد نظر قرار داد، به طور مثال داروی هیدروکسی اوره موجب افزایش HBF میشود. (مسعود وکیلی 1380) به همین علت از آن به عنوان درمان در جهت افزایش HBF و در نتیجه کاهش شدت بتا تالاسمی استفاده میشود. (فیضالله هاشمی گرجی 1389) بعلت شیوع بالای انواع کمخونیهای ارثی آمار دقیقی از میزان بروز هموگلوبینوپاتیها درشهر بندرعباس در دسترس نبوده و به همین علت نیز از این پژوهش میتوان بعنوان ملاکی جهت تصمیمگیری در مورد جلوگیری از انتقال ارثی این قبیل بیماریها و بدنیا آمدن نوزادان مبتلا به انواع تالاسمی استفاده نمود. با تشخیص نوع هموگلوبین نابهنجار و تعیین شیوع آن در جمعیت بومی بندرعباس میتوان به صورت هدفدار آزمایشهای مربوطه را در غربالگری پیش از ازدواج و بقیه موارد که نیاز به شناسایی و غربالگری کمخونی ارثی دارند مد نظر قرار داد. به عنوان نمونه شیوع بیماری فاویسم در استانهای شمالی کشور باعث شده که یکی از آزمایشات درخواستی غربالگری که در این استان در موارد وجود سابقه خانوادگی درخواست میگردد آزمایش G6PD باشد که اولین قدم در شناسایی شیوع بیماری است که در شهرهای مختلف این استانها وجود دارد. روش بررسی :این مطالعه یک پژوهش توصیفی تحلیلی مقطعی است که بر روی کلیه مراجعه کنندگان به بیمارستان خاتمالانبیاء مشروط بر درخواست انجام آزمایشات CBC و الکتروفورز Hb از تاریخ آذر 1392 تا آذر 1393 صورت گرفته است. افراد تحت بررسی در سه گروه سنی 1 تا 20، 20 تا 50، 50 سال به بالا و به دو جنس مرد و زن تقسیم شدند.
ابتدا از همه بیماران آزمایش CBC بعمل آمده و تمام مواردی که طبق برنامه غربالگری کشوری دارای
fl MCV<78 وMCH<27pg بودند جمعآوری و بر روی آنها بعنوان نمونههای صلاحیتدار، آزمایش الکتروفورز با=8.6 PH (قلیایی) انجام شد. نتایج همه نمونهها توسط روش کروماتوگرافی ستونی جهت HbA2 (شرکت تلاشگران)، روش مقاومت قلیایی برایHbF (شرکت بیوژن) و روش حلالیت جهت HbS مورد تأیید قرار گرفتند. در مواردی که تشخیص باند اضافه شکل گرفته برروی ژل الکتروفورز توسط روشهای فوق امکانپذیر نبود اقدام به انجام آزمایش به روش Capillary Electrophoresis با دستگاه CEBIA (Full و یا MINI CAP) گردید.
لازم بذکر است که برای بیماران با HBA2<3.5، آزمایشات آهن و فریتین به عمل آمده و در صورت تأیید فقر آهن، از مطالعه کنار گذاشته شده و در غیر اینصورت در این پژوهش مورد بررسی قرار میگرفتند. بعلت پیچیدگی فیزیولوژیک، زنان باردار از این مطالعه حذف شدهاند.
در مواردی که افراد تحت بررسی سابقه قبلی یا خانوادگی ابتلای به نوع خاصی ازهموگلوبینوپاتی داشتند، بدون توجه به مقادیر MCV و MCH (که در بعضی از موارد بالاتر از معیار برنامه غربالگری کشوری بود) الکتروفورز و در بعضی مواردCapillary Elactrophoresis انجام گرفت که نتایج قابل قبول داشت.
اطلاعات بدست آمده به ترتیب شامل جنس، سن، تعداد RBC، مقادیرHb ،HCT،MCV،MCH،RDW و به ترتیب نوع هموگلوبین (HbA-HbA2-HbF-HbS-HbD.G-HbC.Oarab- آلفا تالاسمی HbH و موارد شناخته نشده) طبقهبندی شده و اطلاعات جمعآوری گشته توسط نرمافزار SPSS version21 مورد ارزیابی قرار گرفت. اطلاعات بدست آمده ابتدا از نظر فراوانی و سپس با استفاده از آزمون آماری
Automatic Linear Modeling Regression مورد بررسی آماری قرار گرفت.
یافتهها:
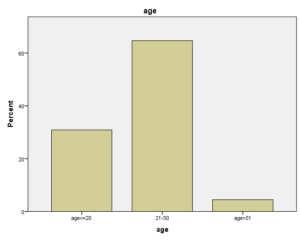
در این مطالعه 5838 نفر که جهت ردیابی هموگلوبینوپاتیهای ارثی به بیمارستان خاتمالانبیاء شهر بندرعباس مراجعه کرده بودند مورد بررسی قرار گرفتند که از میان آنان 85 نفربه دلیل بارداری و 290 نفر بعلت فقر آهن از مطالعه حذف گشته و مطالعه بر روی 5463 نفر باقیمانده صورت پذیرفت که از این تعداد 583 نفر دارای MCV<78fl وMVH<27pg بوده و حائز شرایط لازم برای انجام آزمایش الکتروفورز و بررسی بیشتر بودند. از این تعداد 358 نفر مذکر و 225 نفر مؤنث بودند. 180 نفر در دامنه سنی بین یک الی 20 سال و 377 نفر در دامنه سنی بین 20 الی 50 سال و 26 نفر نیز بالای 50 سال سن داشتند. میانگین تعداد اریتروسیتهای آنان (7104/5) میلیمتر مکعب، حداقل آن با (82/2) و حداکثر آن با (30/8) در میلیمتر مکعب و با میانگین Hb (679/12)، حداقل آن (3/7) و حداکثر آن (5/18) بوده و میانگین HCT آن افراد نیز با (115/39)، حداقل آن (5/21) و حداکثر آن (9/55) بوده و میانگین مقادیرMCV این افراد fl (862/68) و حداقل آن (3/42) فمتولیتر و حداکثر آن (103) فمتولیتر بود که در جدول (1) آمده است.
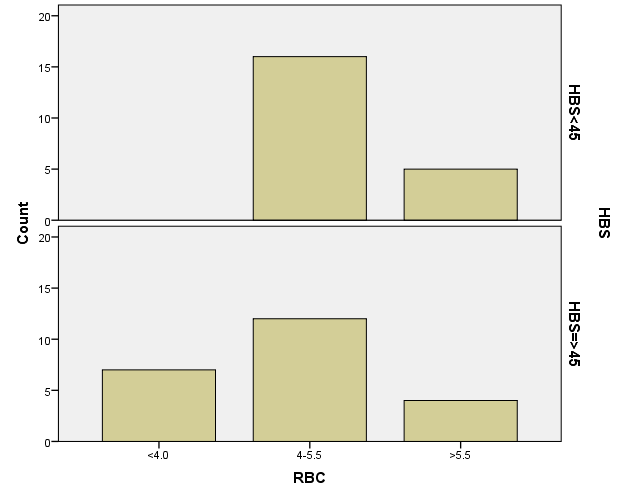
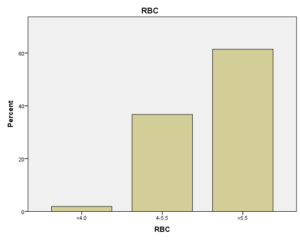
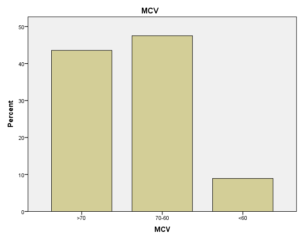
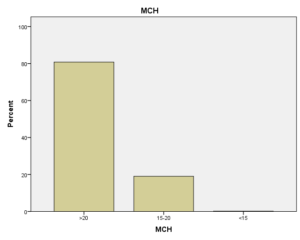
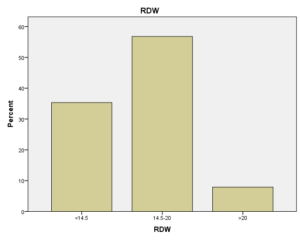
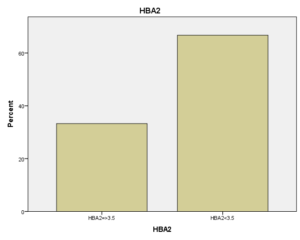
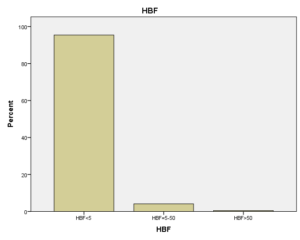
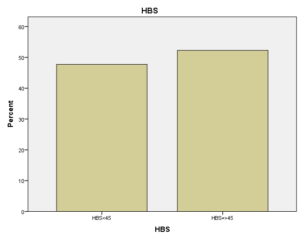
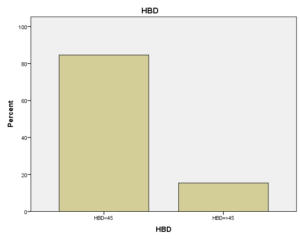
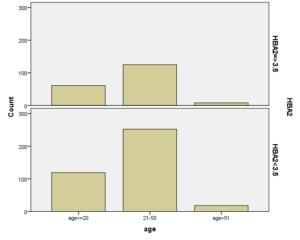
افراد تحت بررسی از نظر HbA2به دو گروه بزرگتر از %3.5 و گروه کوچکتر از %3.5 تقسیم شدند. گروه اول 194 مورد معادل 33.3 % و گروه دوم 389 نفر معادل 66.7% افراد تحت بررسی را شامل میشوند. HbF در یکی از سه گروه، کوچکتر از 5%، از 50-5% و بالاتر از 50% تقسیم شد که به ترتیب 417 نفر که 71.5% و 18 نفر با 3.1% و 2 نفر با 0.3% موارد را شامل میشدند 3.4% میانگین HbF در افراد تحت بررسی بدست آمد. افراد تحت بررسی از نظر HbS به دو گروه کوچکتر از 45% و بزرگتر یا مساوی 45% تقسیم شدند. گروه اول 3.6% موارد و گروه دوم 3.9% موارد را به خود اختصاص داده و 7.5% میانگینHbS در افراد تحت بررسی بدست آمد. HbD نیز به دو گروه کوچکتر از 45% و بزرگتر یا مساوی 45% تقسیم شدند که به ترتیب 55 بیمار در گروه اول با 9.4% موارد و 10 بیمار در گروه دوم با 1.7% موارد قرار داشتند. جمعاً HbD با میانگین 11.1% در این بررسی ثبت شد.HbH وConstant Spring هر کدام با یک مورد 0.2% موارد را به خود اختصاص دادند. از نظر تعداد اریتروسیت در میلیمتر مکعب، افراد تحت بررسی به سه گروه کمتر از چهار میلیون در میلیمتر مکعب، 4 الی 5/5 میلیون در میلیمتر مکعب و بیش از 5/5 میلیون در میلیمتر مکعب تقسیم شدند. در گروه اول 11 نفر معادل 1.9% و گروه دوم 214 نفر معادل 36.7% و گروه سوم 358 نفر معادل 61.4% قرار داشتند. از نظر مقادیر MCV نیز افراد تحت بررسی در سه گروه بیشتر از 70 فمتولیتر، 60 الی 70 فمتولیتر و کمتر از 60 فمتولیتر تقسیم شدند. تعداد افراد هریک از گروههای سهگانه به ترتیب 43.6% و 47.5% و 8.9% موارد را به خود اختصاص دادند. MCH نیز در افراد تحت بررسی در یکی از سه گروه بیشتر از 20 پیکوگرم و 15 الی 20 پیکوگرم و کمتر از 15 پیکوگرم تقسیم شدند که به ترتیب 80.8% و 19% و 0.2% موارد را به خود اختصاص داده بودند. از نظر مقادیر RDW نیز افراد تحت بررسی در سه گروه کمتر از 5/14، 35.3% موارد و 5/14 الی 20، 56.8% و بیشتر از 20، 7.9% موارد تقسیمبندی گردیدند. (همه گرافها در ذیل آمده است)
بحث:
کمخونیهای ارثی در کشور ما و بخصوص در استان هرمزگان و شهر بندرعباس بسیار شایع بوده، (حسینپور فیضی 1388) میتواند ایجاد مشکلات اقتصادی، درمانی و حتی روحی روانی نماید. برنامهریزی جهت تعیین خط مشی نحوه پیشگیری از این قبیل کمخونیها و داشتن آمار درست در رابطه با نوع کمخونی میتواند کمک کننده باشد.
در این پژوهش از 5838 نفری که مورد بررسی قرار گرفتند 583 نفر حامل نوعی هموگلوبین نابهنجار بودند که در بعضی از موارد نه تنها هیچ عارضه بالینی نداشتند بلکه اندکسهای شمارش گلبولی آنها طبیعی و نرمال بود یا بعضی از موارد با تغییرات خفیفی در یکی از فاکتورهای شمارش گلبولی همراه بودند. هیچ یک از افرادی که تحت بررسی قرار گرفتند علائم بالینی واضح نداشتند غیر از افرادی که دچار بیماری سیکل سل هموزیگوت بودند.
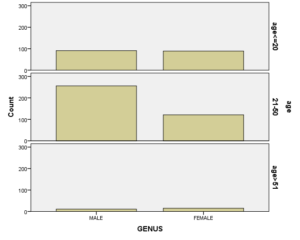
در بین 583 نفری که بررسی شدند 358 نفر مرد و 225 زن بودند. افزایش تعداد مردان نسبت به زنان احتمالاً به این دلیل بود که 375 نفر از افراد تحت بررسی که بیشترشان زنان بودند بدلیل فقر آهن از مطالعه حذف شدند. از لحاظ سنی چون بیشتر کسانی که به بیمارستان خاتمالانبیاء برای درمان بیماری یا انجام تست غربالگری برای تصدی شغل مراجعه کرده بودند در رده سنی بین 20 الی 50 سال قرار داشتند. به همین دلیل بیشتر مراجعه کنندگان ما که 377 نفر معادل 64.7% بودند در این گروه سنی جای داشتند. مقایسه سن و جنس افراد نشان داد که شیوع کمخونی در هر دو جنس در دو گروه سنی زیر 20 سال و بالای 50 سال از لحاظ فراوانی شبیه یکدیگر میباشد. اختلاف در سن 20 تا 50 سال بوده که باروری و شیردهی زنان در این رده سنی باعث بروز فقر آهن در زنان میشود که باعث حذف موارد آن در این بررسی و افزایش درصد مردان میگردد.
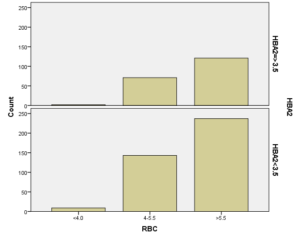
در بررسی گلبول قرمز مشخص شد که بیشترین فراوانی در شمارش گلبول قرمز با 61.4% مربوط به شمارش بالاتر از 5/5 (میلیون در میلیمتر مکعب) بوده، که نشان میدهد که همه انواع هموگلوبینوپاتیهای ارثی (غیر از هموگلوبینوپاتی نوع سیکل سل) به یک نسبت در افزایش شمارش گلبول قرمز مؤثر هستند. (تمام جداول و گرافها در ذیل آمده است):


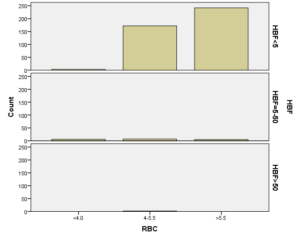
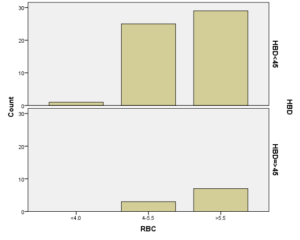
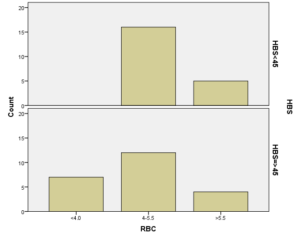
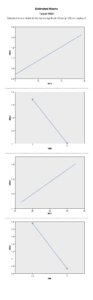
برطبق محاسبات انجام شده مشخص گردید که مهمترین فاکتور پیشبینی کننده وجود هموگلوبینوپاتی (MCV) با 95% و MCH با 77% بوده است. HbA2 نسبت به هر دو فاکتور رابطه عکس داشت یعنی افزایشHbA2 باعث کاهشMCV وMCH با احتمال به ترتیب 95% و 77% شده است. با احتمال بیشتر از 95% نشان داده شده است که افزایش HbA2 با افزایش HbD و HbF همراه بوده است، یعنی هرچه فراوانی HbD وHbF از 45% بیشتر میشود احتمال وجود و همراهیHbA2 نیز بیشتر میشود. پس بیشتر موارد هموگلوبینوپاتیهایD وF با ژن تالاسمی بتا نیز همراه هستند. (به شکل ذیل توجه نمایید)
نکته جالب در بررسی شیوع و فراوانی هموگلوبینوپاتی نسبت به سن این است که شیوع انواع هموگلوبینوپاتی در هر سه رده سنی زیر 20 سال (سنی که اغلب پدر و مادرهای این افراد بعد از انجام آزمایشهای غربالگری قبل از ازدواج و جستجوی موارد هموگلوبینوپاتیها و مشاوره با هم ازدواج کردند)، سن 20 الی 50 سال و سن بالای 50 سال به طور معنیداری شبیه به هم بوده است. این آزمون نشان میدهد غربالگری قبل از ازدواج در جلوگیری از شیوع انواع هموگلوبینوپاتیها کارایی لازم را نداشته است.
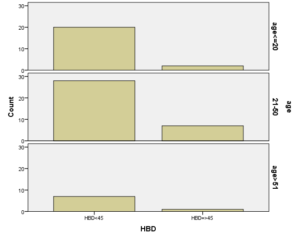
نتایج حاصل از این مطالعه با توجه به شیوع بالای سیکلسل در این شهر نتایج مطالعات دکتر عابدیان در شمال کشور را تأیید میکند که افراد مبتلا به آنمی داسی عمدتاً الگوی ویژهای دارند بطوریکه در حالت Sickel Trait داراي MCV بالاتر از 80 و HBA2 كمتر از3.5% و مقدار HBS 30-40% ميباشند. افراد مبتلا به Sickel Thal داراي MCV كمتر از 70، HBA2 بيشتر از 3.5% و HBS ،50-80% بودند. برخلاف بقیه نقاط کشور در بین 538 نفری که مورد بررسی قرار گرفتند مقدار آلفا تالاسمی 43.1% بود. حدوداً معادل 4% آلفا تالاسمی و 5% بتا تالاسمی در کل جمعیت مورد بررسی در این شهر ثبت گردید. یکی دیگر از موارد مهم در این شهر وجود 1% HbD در کل جمعیت مورد بررسی و 1% HbS میباشد که در همانتقالی با بتا تالاسمی به علت شیوع بالا هر دو میتواند باعث افزایش شدت و تظاهرات بالینی کمخونی شود.
بررسی تعیین جهشهای هموگلوبین D به روش مولکولی در مازندران که توسط پیام روشن انجام گرفت نشان داد که جهشها باعث ایجاد انواع مختلف HbD میشوند که میتواند در شرایط بالینی مختلف تظاهرات غیر یکسان از خود نشان بدهد. در این مطالعه موارد متعددی دیده شده است که دو بیمار با HbD مشابه، اندکسهای بسیار متفاوتی از خود نشان دادند همانطور که در شکل بالا مشاهده نمودید این بیمار طبق برنامه غربالگری کشوری جزء موارد هموگلوبینوپاتی محسوب نمیشود پس لازم است برای تمام افراد دارای سابقه خانوادگی برای جستجوی HbD ابتدا روش الکتروفورز انجام شود و نوع HbD افراد حتماً به روش ملکولی تعیین شود.
نتیجهگیری:
شیوع هموگلوبینوپاتی در بندرعباس 9.4% میباشد که 4%آن آلفا تالاسمی است، بنابراین در شهر بندرعباس HbA2 ملاک مناسبی برای بررسی هموگلوبینوپاتی نیست و چون در این استان مثل اکثر نقاط کشور امکانات مولکولی بررسی کمخونی ارثی وجود ندارد باید در صورت وجود کمخونی وHBA2<3.5 حتماً فقر آهن بررسی شده و در صورت نبود فقر آهن با بودن MCV<78 و MCH<27، بیمار به عنوان آلفا تالاسمی در نظر گرفته شود دوم اینکه برای بررسی کمخونی ارثی به اندکسهای CBC اعتماد نکرده و حتماً الکتروفورز انجام شود چون بعضی انواع هموگلوبینوپاتی روی اندکس CBC تأثیر نمیگذارند.
همانطور که ملاحظه کردید شیوع هموگلوبینوپاتی در رده سنی مختلف یکسان بوده و با توجه به اینکه ایران روی کمربند جغرافیایی تالاسمی قرار دارد برطبق بررسیهای انجام شده ازدواج فامیلی در هرمزگان 38٪ است و 27٪ آنها در میان عموزادهها و عمهزادهها که زادههای نخست هستند میباشد (حسینپور فیضی 1388) پیامد آن در خزانه ژنی موجب افزایش بروز اشکال شدید و پیچیده و یا بسیار خفیف و بدون علایم نموده است (حسینپور فیضی.1388) پس بالاجبار وقتی ازدواج فامیلی بسیار خطرساز است حتماً در ازدواج فامیلی در این شهر مشاوره و آگاهی بیشتری راجع به این بیماریها داده شود و برای تمام زوجین در معرض خطر بدنیا آوردن نوزاد بیمار تالاسمی بتا ماژور و یا سایر هموگلوبینوپاتیهایی که باعث بروز بیماری میشوند، مراکزی دائر شود که بتوانند با اطمینان بالایی بیماری را در نوزادان قبل از تولد تشخیص دهند تا به رفع این مشکل کمک شود.
منابع:
1- دکتر خدامراد زندیان 1381 مجله دانشگاه علوم پزشکی اهواز (ارزیابی شاخصهای گلبول قرمز در بتا تالاسمی مینور)
2- بررسی پارامترهای هماتولوژیک و سطح هموگلوبین A2 و F در والدین بیماران مبتلا به تالاسمی ماژور در بیرجند در سال 1381 طیبه چهکندی مجله دانشگاه علوم پزشکی سبزوار
| 3- ارتباط هليكوباكترپيلوري با ميزان هموگلوبين و MCV- Mean Corpuscular Volume و فرتين سرم در بيماران كمخون
مراجعه كننده به كلينيك فوق تخصصي خون و انكولوژي از اول آبان 1380 الي پايان دي 1381 در شهر اراک. مشفقی کامران، رفیعی محمد |
| 4-بررسي تغييرات سطح خوني كربواكسي هموگلوبين و غلظت پلاسمايي ليپيد و ليپوپروتئين در كودكان قرار گرفته
در معرض دود سيگار |
| رهباني محمدابراهيم*، رحيمي پورعلي |
| * گروه كودكان، بيمارستان كودكان، دانشگاه علوم پزشكي تبريز |
5- اهمیت اندازهگیری HBF در تشخیص افتراقی کمخونی داسی شکل از سیکل بتا تالاسمی، پورفتحالله علیاکبر
6- بررسی درصد هموگلوبین A2 در ناقلین قطعی بتا تالاسمی مینور مراجعه کننده به مرکز درمانی بیماریهای خاص کرمان، دکتر منیژه عطاپور مجله دانشگاه علوم پزشکی کرمان
| 7- مقايسه روشهاي مختلف شناسايي آلفا تالاسمي با روش بررسي زنجيره زتا از طريق الكتروفورز زنجيرههاي هموگلوبين |
| پاسالار پروين، خالقيان مليحه، شريفيان رمضانعلي |
8- بررسی تأثیر هیدروکسی اوره بر میزان تولید هموگلوبین جنینی و فراوانی حملات دردناک در کمخونی داسی شکل
دکتر مسعود وکیلی 1380 مجله دانشگاه علوم پزشکی ایران
9- بیماری هموگلوبین H، دکتر خدامراد زندیان 1379
10- شیوع هموگلوبینوپاتی با توجه به میزان انواع هموگلوبین و ارتباط آن با حجم متوسط گلبولی در بین دانشآموزان پیش دانشگاهی شهر بوشهر، دکتر علی موحد مرکز تحقیقات طب گرمسیری و عفونی خلیج فارس 1388
11- بررسی ارتباط افزایشی mir-210 و بیان زنجیره گاما، دکتر سید امیر حسین امامی، مجله دانشکده پیراپزشکی دانشگاه علوم پزشکی تهران 1390
12- شیوع هموگلوبینوپاتی در استان سیستان و بلوچستان در جنوب شرق ایران، ابراهیم میری مقدم 1391 دانشگاه علوم پزشکی زاهدان
13-Sickle cell retinopathy: diagnosis and treatment Bonanomi MT, Lavezzo MM.2012
14-Screening immediate family members for carrier identification and counseling: a cost-effective and practical approach.
Ansari SH, Baig N, Shamsi TS, Saif-ur-Rehman, Ansari ZH, Behar Z, Perveen K, Erum S, Bukhari ZR, Khan MT, AkBar M.
- Department of Hematology, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. muddasirsaqiB@yahoo.com
- 15- Identification of one or two α-globin gene deletions By isoelectric focusing electrophoresis.
- Agarwal AM, Nussenzveig RH, Hoke C, Lorey TS, Greene DN.
16-Five Hemoglobin Variants in a Double Heterozygote for α- and β-Globin Chain Defects.
Singha K, Fucharoen G, Fucharoen S.
17-Analysis of hemoglobin electrophoresis results and physicians investigative practices in Saudi AraBia.
Pathology, Era’s Lucknow Medical College, Lucknow, Uttar Pradesh, India. Department of
18-HemogloBinopathies in South Gujarat population and incidence of anemia in them
Ankur G. Patel, Avani P. Shah, Smita M. Sorathiya, and Snehalata C. Gupte
Author information ► Copyright and License information ►
19-Assessment of the red cell proteome of young patients with unexplained hemolytic anemia By two-dimensional differential in-gel electrophoresis (DIGE).
von Löhneysen K, Scott TM, Soldau K, Xu X, Friedman JS.
20-Analysis of hemoglobin electrophoresis results and physicians investigative practices in Saudi Arabia
Syed Riaz Mehdi and Badr Abdullah Al Dahmash
21-Abnormal haemoglobins: detection & characterization
Henri Wajcman and Kamran Moradkhani*
22- هماتولوژی هنری دیویدسون 2006
23- دکتر اصنافی، شیوع کمخونی و ارتباط آن با بارداری 1382-
24- دکتر جعفر نصوحی، میزان افت هموگلوبین در اعمال جراحی زنان و زایمان 1382
25- مهدی حقی- ناصر پولادی- محمدعلی حسینپور فیضی- عباسعلی حسینپور فیضی، بتا تالاسمی در ایران 1389
26- محمدرضا کرامتی 1388، تعیین مقادیر خونشناسی در نوزادان شهر مشهد
27- بهناز زربخش، بررسی مولکولی آلفا تالاسمی از نظر جهشهای حذفی و غیرحذفی در حاملین مشکوک ایرانی
1389
28- الهام ایرانی، مقایسه کیفیت زندگی بیماران مبتلا به تالاسمی ماژور بر اساس شرکت در فعالیتهای گروهی در شهرستان بندرعباس 1392
29-پیام روشن- دکتر محمدرضا مهدوی- دکتر محمدباقر هاشمی سوته- محمدطاهر حجتی- نسیم یوسفیان 1392، تعیین جهشهای هموگلوبین به روش مولکولی در مازندران
ورود / ثبت نام