
متابولیسم پروتئینها و اسیدهای آمینه و اختلالات ناشی از آنها
مراد رستمي: کارشناس ارشد بیوشیمی بالینی، دانشگاه علوم پزشكي جندیشاپور اهواز
معصومه جرفی: کارشناس ارشد میکروبشناسی، دانشگاه علوم پزشكي جندیشاپور اهواز
اختلالات ناشی از متابولیسم اسیدهای آمینه
هضم و جذب پروتئینها:
آنزیمهای پروتئولیتیک (پروتئازها) موجب تجزیه پروتئینهای رژیم غذایی به اسیدهای آمینه تشکیلدهنده آنها در معده و روده میشوند. بسیاری از این پروتئازهای هضمکننده بهصورت اشکال بزرگتر و غیرفعال، موسوم به زیموژن ساخته میشوند که پس از ترشح به مجرای گوارش، شکسته شده و تولید پروتئازهای فعال را مینمایند.
در معده، پپسین موجب هضم پروتئینها از طریق هیدرولیز آنها به پلیپپتیدهای کوچکتر میشود، سپس، محتویات معده وارد روده کوچک میشود. در روده کوچک، آنزیمهای تولیدشده توسط بخش اگزوکراین پانکراس، عمل میکنند. پروتئازهای پانکراس (تریپسین، کیموتریپسین، الاستاز و کربوکسی پپتیدازها) موجب تجزیه پلیپپتیدها به الیگوپپتیدها و اسیدهای آمینه میشود.
تجزیه بیشتر الیگوپپتیدها به اسیدهای آمینه توسط آنزیمهای تولیدشده توسط سلولهای اپیتلیال روده کوچک، کامل میشود. این آنزیمها شامل آمینوپپتدازها (که در لبه برسی قرار دارند) و سایر پپتیدازها (که در درون سلولها قرار دارند) میباشند. سرانجام، اسیدهای آمینه تولیدشده از هضم پروتئینها، جذب سلولهای اپیتلیال روده کوچک شده و وارد گردش خون میگردند.
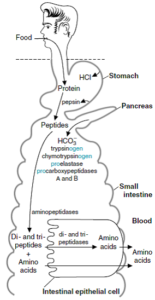
پپسین به شکل پپسینوژن از سلولهای اصلی معده ترشح شده و در pH اسیدی معده و یا به طریق اتوکاتالیتیک (توسط خود پپسین)، به شکل فعال تبدیل میگردد. تریپسین، کیموتریپسین، الاستاز و کربوکسی پپتیدازها نیز به شکل غیرفعال توسط پانکراس به داخل روده باریک ترشح میشوند. تبدیل تریپسینوژن به تریپسین توسط آنتروپپتیداز (آنتروکیناز) مخاط دوازدهه و یا به طریق اتوکاتالیتیک (توسط خود تریپسین) صورت گرفته و سپس تریپسین سبب فعالسازی کیموتریپسینوژن، پروالاستاز و پروکربوکسی پپتیدازها میگردد. آمینوپپتیدازها در سطح مجرایی سلولهای اپیتلیال روده و دیپپتیدازها در داخل این سلولها وجود دارند.
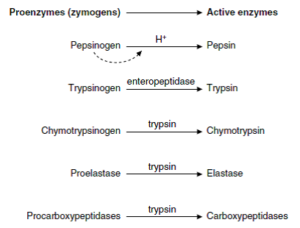
فعالسازی زیموژنهای معدی و پانکراتیک
انتقال اسیدهای آمینه:
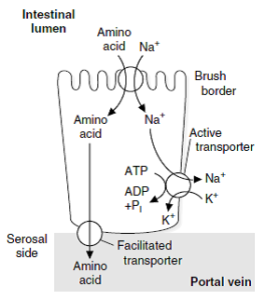
انتقال اسیدهای آمینه در سلولهای اپیتلیال روده
حاملهای وابسته به سدیم، موجب انتقال سدیم و اسیدهای آمینه از لومن روده به سلولهای اپیتلیال روده میشوند (انتقال فعال ثانویه). Na+ به سمت سروزال (از طریق غشاء بازولترال) و در تعویض با K+ توسط پمپ – ATPase Na+,K+، پمپ میشود. در سمت سروزال، اسیدهای آمینه توسط انتشار تسهیل شده و در جهت گرادیان غلظتی، وارد خون میشود.
شباهت زیادی بین مکانیسمهای انتقال اسیدهای آمینه در روده و جذب گلوکز وجود دارد. دیپپتیدها در داخل سلولهای اپیتلیال ابتدا به اسیدهای آمینه تجزیه شده و سپس جذب گردش خون میشوند. در چند روز ابتدایی زندگی، احتمال جذب پروتئینها به طریق آندوسیتوز وجود دارد. برای انتقال اسیدهای آمینه در سلولها، دستگاههای انتقالی مختلفی وجود دارد. برخی از این دستگاهها بهصورت انتقال تسهیل شده و برخی دیگر توسط انتقال فعال وابسته به سدیم، عمل میکنند.
انتقال اسیدهای آمینه از عرض غشاهای سلولی برای جذب گوارشی و بازجذب آنها در کلیهها مهم است. در اغلب موارد، مکانیسمهای انتقالی این دو عضو، مشابه بوده و اختلال در یک سیستم انتقالدهنده همراه با دفع گوارشی و کلیوی یک و یا چند تا از اسیدهای آمینه است. تنها در دو مورد (سوءجذب متیونین و تریپتوفان)، مشکل فقط در مخاط روده بهتنهایی وجود دارد.
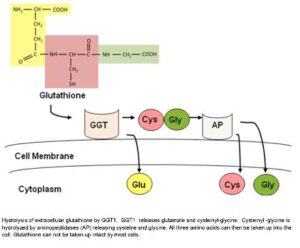
در برخی از بافتها، برای انتقال اسیدهای آمینه از عرض غشاء، از چرخه گاماگلوتامیل استفاده میشود. در این چرخه، ابتدا آنزیم GGT موجب انتقال ریشه گاماگلوتامیل از گلوتاتیون به اسیدآمینه شده و بدینترتیب دیپپتید حاصل، از غشاء عبور مینماید. این دیپپتید در داخل سلول، به اسیدآمینه و اکسوپرولین تجزیه میشود.
تخریب پروتئینهای داخلی:
روزانه حدود 1% پروتئینهای بدن، بهویژه در عضلات، تجزیه شده و مجدداً مورد استفاده قرار میگیرند (نوسازی پروتئینی). پروتئولیز از 2 طریق صورت میگیرد:
1- پروتئولیز وابسته به ATP: پروتئینهای ناقص و دارای عمر کوتاه از این مسیر تجزیه میشوند. در این مسیر، نیاز به یوبیکوئیتین وجود دارد. وجود توالی PEST (پرولین، گلوتامات، سرین و ترئونین)، نشانهای برای اتصال یوبیکوئیتین و تجزیه سریع است. تخریب در داخل پروتئازوم صورت میگیرد.
2- پروتئولیز غیروابسته به ATP: پروتئینهای خارج سلولی، پروتئینهای غشایی و پروتئینهای داخل سلولی دارای عمر زیاد از این مسیر تجزیه میشوند. پروتئینهای خارج سلولی توسط مکانیسم آندوسیتوز توسط سلولهای کبدی برداشت میشوند. تخریب در داخل لیزوزومها صورت میگیرد.
بیوسنتز اسیدهای آمینه:
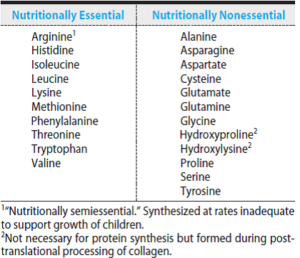
اسیدهای آمینه ضروری: توسط بدن قابل سنتز نبوده و باید از طریق رژیم غذایی وارد بدن شوند.
اسیدهای آمینه غیرضروری: به مقادیر کافی در بدن قابل سنتز هستند. Arg توسط بدن قابل سنتز هست، اما ظرفیت بدن برای سنتز آن پایین است (نیمه ضروری)، از این رو، در مواقع افزایش نیاز (خانمهای باردار و بچههای در حال رشد)، باید از طریق رژیم غذایی تأمین شود.
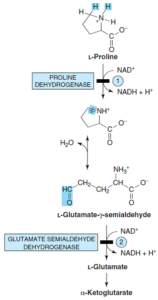
پرولین
پرولین به جای ترانسآمیناسیون مستقیم، ابتدا به دهیدروپرولین اکسید شده که آن نیز با گرفتن آب به گلوتامات γ- سمیآلدئید تبدیل میشود. این ترکیب سپس به گلوتامات، اکسید و به α- کتوگلوتارات، ترانسآمینه میشود.
دو نوع هیپرپرولینمی اتوزومی مغلوب، شناسایی شده است:
1- هیپرپرولینمی نوع 1: نقص در پرولین دهیدروژناز وجود دارد. بیماری در هتروزیگوتهای هیپرپرولینمی نوع 1 خفیف است. در این نوع از هیپرپرولینمی، اختلال کاتابولیسم هیدروکسی پرولین وجود ندارد. این بیماران دارای هیپرپرولینمی هستند.
2- هیپرپرولینمی نوع 2: نقص در گلوتامات γ- سمیآلدئید دهیدروژناز وجود دارد. در این نوع از اختلال، کاتابولیسم پرولین و هیدروکسی پرولین مختل است. ادرار این بیماران حاوی یکی از کاتابولیتهای هیدروکسی پرولین موسوم به 1∆- پرولین 3- هیدروکسی 5- کربوکسیلات میباشد. این بیماران بر خلاف هتروزیگوتهای نوع 1 فاقد هیپرپرولینمی هستند.
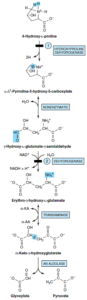
3- 4- هیدروکسی پرولین: 4- هیدروکسی L- پرولین توسط یکی از دهیدروژنازهای میتوکندری به L- 1∆- پرولین 3- هیدروکسی 5- کربوکسیلات اکسید میشود که در حالت تعادل غیرآنزیمی با γ- هیدروکسی L- گلوتامات γ- سمیآلدئید است. سمیآلدئید به اریترو γ- هیدروکسی L- گلوتامات اکسید شده و سپس به α- کتو γ- هیدروکسی گلوتارات ترانسآمینه میشود، سپس با نوعی شکست آلدولی، گلیاگزیلات و پیروات ساخته میشود.
4- هیپرهیدروکسی پرولینمی: هیپرهیدروکسی پرولینمی، یک صفت اتوزومی مغلوب با نقص در 4- هیدروکسی پرولین دهیدروژناز است و با میزان بالای 4- هیدروکسی پرولین در پلاسما مشخص میشود. هیچ اختلالی از کاتابولیسم پرولین همراه آن نیست، زیرا آنزیم مبتلا فقط در کاتابولیسم هیدروکسی پرولین نقش دارد. این وضعیت، هیچ تأثیری بر متابولیسم کلاژن نداشته و ظاهراً همانند هیپرپرولینمیها، خوشخیم میباشد.
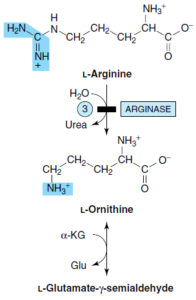
آرژینین
از کاتابولیسم آرژینین، α- کتوگلوتارات حاصل میشود. آرژینین تحتتأثیر آرژیناز و شکسته شدن هیدرولیزی گروه گوانیدین به اورنیتین تبدیل میشود. سپس گروه 5- آمین اورنیتین دستخوش ترانسآمیناسیون شده و تولید گلوتامات γ- سمیآلدئید مینماید که در ادامه، همانند مسیر کاتابولیسم پرولین، به α- کتوگلوتارات تبدیل میشود.
دو اختلال ارثی وجود دارند که منجر به هیپرارنیتینمی میشوند:
1- آتروفی حلقوی (Gyrate atrophy) شبکیه: نقص در ارنیتین δ- ترانسآمیناز وجود دارد. این صفت اتوزومی مغلوب به صورت دژنرسانس مشیمیه (کورویید) و شبکیه به همراه افت فزاینده دید محیطی، دید تونلی و در نهایت، کوری تظاهر مینماید. در این اختلال، سطح اورنیتین پلاسما افزایش مییابد. درمان شامل محدودسازی آرژینین رژیم غذایی میباشد.
2- سندروم هیپرارنیتینمی- هیپرآمونمی: احتمالاً نقص در نوعی ناقل تبادلی ارنیتین- سیترولین وجود دارد و از این رو، این سندروم را میتوان نوعی نقص چرخه اوره به حساب آورد. این اختلال ژنتیکی مغلوب، به دلیل اختلال در انتقال اورنیتین به درون میتوکندری، با میزان بالای اورنیتین (هیپرارنیتینمی) و آمونیاک خون (هیپرآمونمی)، همراه میباشد.
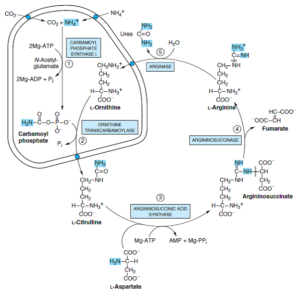
هیستیدین
هیستیدین تحتتأثیر هیستیداز، دزآمیده شده و تولید اوروکانات مینماید. سپس اوروکانات با گرفتن H2O و انجام نوعی اکسید و احیای داخلی تحتتأثیر اوروکاناز به 4- ایمیدازولون- 5- پروپیونات تبدیل میشود. این ترکیب با هیدرولیز به N- فرمیمینوگلوتامات (فیگلو) تبدیل میگردد. گروه فرمیمینوی فیگلو به تتراهیدروفولات، منتقل شده و گلوتامات ساخته میشود. سپس گلوتامات با ترانس آمیناسیون به α- کتوگلوتارات تبدیل میشود.
در کمبود اسید فولیک، واکنش تبدیل N- فرمیمینوگلوتامات (فیگلو) به گلوتامات، به طور نسبی و یا کامل متوقف شده و فیگلو از طریق ادرار دفع میگردد، بنابراین، دفع فیگلو در ادرار، متعاقب تجویز یک دوز آزمایشی هیستیدین، یکی از تستهای تشخیصی کمبود اسید فولیک میباشد.
افزایش زیاد دفع هیستیدین، از یافتههای معمولی در بارداری طبیعی است که به علت تغییری موقت در عملکرد کلیه میباشد.
دو نوع اختلال ظاهراً خوشخیم در کاتابولیسم هیستیدین شناسایی شده است:
1- هیستیدینمی: نقص در هیستیداز وجود دارد و لذا تبدیل هیستیدین به اوروکانات مختل میشود. این سندروم با میزان بالای هیستیدین در خون و ادرار مشخص شده و در اکثر افراد خوشخیم میباشد.
2- اوروکانیک اسیدوری: نقص در اوروکاناز وجود دارد و بنابراین، موجب ازدیاد دفع اوروکانات میشود. یک اختلال اتوزومی مغلوب است. دفع زیاد اسید اوروکانیک، تنها نشانه این اختلال ظاهراً خوشخیم میباشد.
اختلالات مربوط به ترکیبات مرتبط با هیستیدین:
1- کمبود کارنوزین:
کارنوزین توسط کارنوزیناز (کارنوزین هیدرولاز) به β-آلانین و L-هیستیدین هیدرولیز میشود. اختلال ارثی کمبود کارنوزین با کارنوزینوری مشخص میشود.
2- هموکارنوزینوز: کارنوزین سنتتاز وظیفه ساخت هموکارنوزین در بافت مغز را برعهده دارد. کارنوزیناز سرم، هموکارنوزین را هیدرولیز نمیکند. هموکارنوزینوز یک اختلال فوقالعاده نادر ژنتیکی است که احتمالاً به علت کمبود کارنوزیناز سرم ایجاد میشود. این اختلال با عقبماندگی ذهنی و پاراپلژی اسپاسمی فزاینده همراه است. پاراپلژی، یک اختلال در عملکرد حرکتی یا حسی اندامهای تحتانی میباشد.
3- 3- متیل هیستیدین: 3- متیل هیستیدین در ادرار مبتلایان به بیماری ویلسون به طور غیرعادی پایین است. غلظت آن در ادرار طبیعی، حدود mg/dl 50 میباشد.
گلیسین
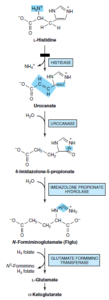
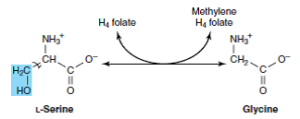
گلیسین میتواند به سرین و سپس به پیروات تبدیل شود.

در انسان و بسیاری از مهرهداران دیگر، مسیر اصلی کاتابولیسم گلیسین و سرین، احتمالاً شکست گلیسین میباشد. کمپلکس گلیسین سنتاز، یک کمپلکس آنزیمی ماکروملکولی در میتوکندریهای کبد است که گلیسین را به CO2 و NH4+ میشکند و N5، N10– متیلن تتراهیدروفولات را به صورت برگشتپذیر میسازد.
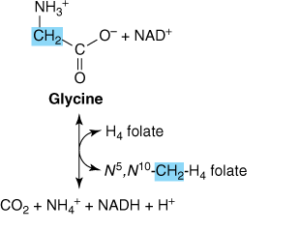
دو نوع اختلال در کاتابولیسم گلیسین شناسایی شده است:
1- گلیسینوری: گلیسینوری با دفع روزانه 1-0/6 گرم گلیسین در ادرار و تمایل به ایجاد سنگهای ادراری اگزالاتی مشخص میشود. در این اختلال، میزان گلیسین پلاسما طبیعی بوده و از این رو، احتمالاً نقص در بازجذب توبولهای کلیه وجود دارد.
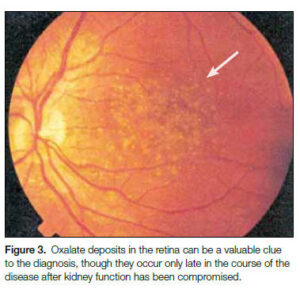
2- هیپراگزالوری اولیه: دفع ادراری اگزالات در هیپراگزالوری اولیه (Primary hyperoxaluria) ارتباطی با دریافت غذایی اگزالات ندارد. گلیسین با دزآمیناسیون به گلیاگزیلات و سپس به اگزالات تبدیل میشود. نقص متابولیک در این بیماری، به صورت ناتوانی در کاتابولیزه کردن گلیاگزیلات و اکسید شدن آن به اگزالات میباشد. در این اختلال، تشکیل فزاینده سنگهای کلیوی دوطرفه با اگزالات کلسیم، نفروکلسینوز و عفونت مکرر دستگاه ادراری باعث نارسایی کلیه یا هیپرتانسیون و در نهایت مرگ زودهنگام میشود.
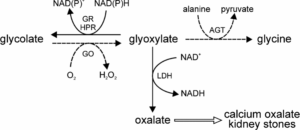
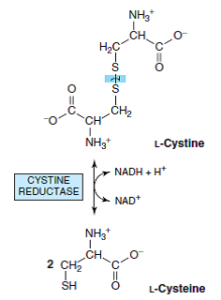
سیستین
در پستانداران، سیستین تحتتأثیر سیستین ردوکتاز به سیستئین تبدیل میشود.
سیستئین:
سیستئین از دو مسیر کاتابولیزه میشود:
1- مسیر مستقیم اکسیداتیو (مسیر سیستئین سولفینات)
2- مسیر ترانسآمیناسیون (مسیر 3- مرکاپتو پیروات)
سیستئین توسط سیستئین دیاکسیژناز و با کمک Fe2+ و NAD(P)H به سیستئین سولفینات تبدیل میشود. کاتابولیسم بیشتر سیستئین سولفینات احتمالاً شامل ترانسآمیناسیون آن به β- سولفینیل پیروات میباشد. تبدیل β- سولفینیل پیروات به پیروات و سولفیت که با دسولفیناز کاتالیز میشود، حتی در غیاب کاتالیز آنزیمی نیز صورت میگیرد.
ترانسآمیناسیون برگشتپذیر سیستئین به 3- مرکاپتوپیروات (تیول پیروات) به وسیله نوعی سیستئین ترانسآمیناز خاص یا با گلوتامات یا آسپارژین ترانسآمینازهای کبد و کلیه پستانداران انجام میشود. احیای 3- مرکاپتوپیروات توسط L- لاکتات دهیدروژناز منجر به تشکیل 3- مرکاپتولاکتات میشود که به صورت دیسولفید مختلط با سیستئین در ادرار طبیعی انسان وجود دارد و در بیماران دچار مرکاپتولاکتات سیستئین دیسولفیدوری به مقدار زیاد در ادرار دفع میگردد. مسیر دیگر، دسولفوره شدن 3- مرکاپتوپیروات به پیروات و H2S میباشد.
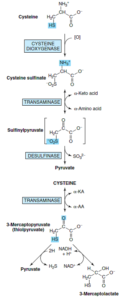
نقایص مربوط به مسیر کاتابولیسم سیستئین شامل موارد زیر میباشند:
1- سیستینوری (سیستین- لیزینوری): در این بیماری، دفع ادراری سیستئین تا 30 برابر حد طبیعی مشخص میشود. در این اختلال، دفع ادراری لیزین، آرژینین و اورنیتین هم افزایش مییابد و در نتیجه ممکن است در مکانیسمهای بازجذب کلیوی این 4 اسیدآمینه، نقصی وجود داشته باشد. از این رو، برخی منابع نام سیستین- لیزینوری را برای این اختلال، مناسبتر میدانند. به دلیل نامحلول بودن نسبی سیستین، در توبولهای کلیوی بیماران سیستینوریک، سنگهای سیستینی تشکیل میشود. دیسولفید مختلط L- سیستئین و L- هموسیستئین که در ادرار بیماران سیستینوریک وجود دارد، محلولتر از سیستین بوده و لذا تشکیل بلورها و سنگهای سیستینی را کاهش میدهد.
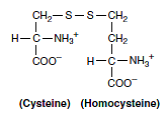
2- سیستینوز (بیماری ذخیرهای سیستین): سیستینوز نوعی اختلال نادر لیزوزومی است که با نقص انتقال سیستین با حامل مشخص میگردد. بلورهای سیستین در بافتها و اعضا و به ویژه در دستگاه رتیکولواندوتلیال رسوب میکنند. سیستینوز اغلب با آمینواسیدوری عمومی (Generalized aminoaciduria) همراه است. سایر اعمال کلیوی هم بشدت مختل شده و بیماران اغلب در سنین جوانی بر اثر نارسایی حاد کلیه فوت میکنند.
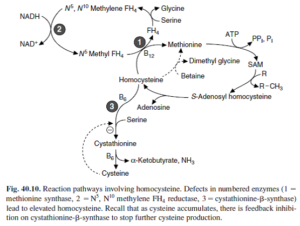
3- هموسیستینوریها: مبتلایان به این نقایص ارثی کاتابولیسم متیونین، روزانه تا 300 میلیگرم هموسیستئین از ادرار دفع میکنند که گاه با S- آدنوزیل متیونین نیز همراه میباشد. سطح متیونین پلاسما نیز بالا میباشد. 4 نقص متابولیک باعث هموسیستینوری میشوند که شامل موارد زیر میباشند:
- هموسیستینوری I: نقص در سیستاتیونین β- سنتاز
- هموسیستینوری II: نقص در N5، N10– متیلن تتراهیدروفولات ردوکتاز
- هموسیسینوری III: میزان کم N5– متیل تتراهیدروفولات- هموسیستئین ترانسمتیلاز به علت ناتوانی در ساخت متیلکوبالامین
- هموسیستینوری IV: میزان کم N5– متیل تتراهیدروفولات- هموسیستئین ترانسمتیلاز به علت نقص در جذب رودهای کوبالامین
یافتههای بالینی در هموسیستینوری نوع I شامل ترومبوز، استئوپورز، دررفتگی عدسی چشم و غالباً عقبماندگی ذهنی میباشد. هر دو شکل پاسخدهنده به ویتامین B6 و مقاوم به ویتامین B6 شناسایی شدهاند. اگر بیماری در اوایل زندگی آغاز شود، رژیم غذایی کم متیونین و پر سیستئین میتواند مانع از تغییرات مرضی شود. سایر انواع هموسیستینوری ناشی از نقص در چرخه متیلاسیون مجدد میباشند.
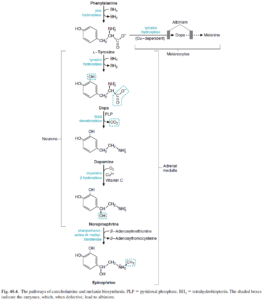
فنیلآلانین
فنیلآلانین توسط فنیلآلانین هیدروکسیلاز به تیروزین تبدیل میشود.
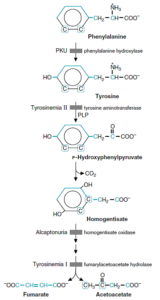
اختلالات متابولیک کاتابولیسم فنیلآلانین، شامل موارد زیر میباشد:
1- فنیل کتونوری کلاسیک (PKU): در این اختلال، فقدان فنیلآلانین هیدروکسیلاز وجود داشته و عقبماندگی ذهنی، عارضه اصلی آن میباشد. سایر علائم بالینی این اختلال شامل تشنج، روانپریشی (Psychosis)، اگزما و نوعی «بوی موشی» میباشد.
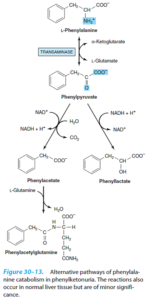
از آن جایی که این بیماران، قادر به تبدیل فنیلآلانین به تیروزین نمیباشند، کاتابولیتهای دیگر یعنی فنیللاکتات، فنیلاستات و فنیلاستیل گلوتامین تولید مینمایند. بخش زیادی از فنیلاستات به صورت فنیلاستیل گلوتامین دفع میگردد. با تجویز رژیم غذایی حاوی مقادیر کم فنیلآلانین میتوان جلوی زوال عقلی کودکان فنیلکتونوریک را گرفت. این رژیم را میتوان در 6 سالگی قطع نمود، زیرا در این زمان، غلظتهای زیاد فنیلآلانین و مشتقات آن دیگر به مغز آسیب نمیرسانند.
غربالگری PKU در برخی از کشورها در مورد نوزادان انجام میشود. از آن جا که دریافت پروتئین غذایی در روزهای اول تولد کم است، ممکن است میزان فنیلآلانین خون شیرخواران فنیلکتونوریک تا روز سوم یا چهارم حیات، بالا نرود. در شیرخواران نارس ممکن است به علت تأخیر در تکامل آنزیمهای کاتابولیسم فنیلآلانین، نتایج مثبت کاذب ایجاد شود. یکی از آزمایشهای مفید غربالگری که اعتبار کمتری دارد، کشف میزان بالای فنیلپیروات ادراری به کمک کلریدفریک است.
2- هیپرفنیلآلانینمی ماندگار: در این اختلال، کاهش فنیلآلانین هیدروکسیلاز وجود دارد.
3- هیپرفنیلآلانینمی خفیف موقت: در این اختلال، تأخیر در تکامل فنیلآلانین هیدروکسیلاز وجود دارد.
4- کمبود دیهیدروبیوپترین ردوکتاز: در این اختلال، کمبود یا فقدان دیهیدروبیوپترین ردوکتاز وجود دارد.
5- اختلال عمل دیهیدروبیوپترین: در این اختلال، نقص ساخت دیهیدروبیوپترین ردوکتاز وجود دارد.
6- هیپرفنیلآلانینمی و تیروزینمی ماندگار: در این اختلال، کاتابولیسم تیروزین نقص دارد.
7- تیروزینمی موقت نوزادی: در این اختلال، مهار p- هیدروکسی فنیل پیروات اکسیداز وجود دارد.
8- تیروزینمی ارثی: در این اختلال، کمبود p- هیدروکسی فنیل پیروات اکسیداز، تیروزین آمینوترانسفراز سیتوپلاسمی و فوماریل استواستات هیدرولاز وجود دارد.
تیروزین
متابولیسم تیروزین شامل موارد زیر میباشد:
1- تبدیل تیروزین به p– هیدروکسی فنیل پیروات: از طریق ترانسآمیناسیون و توسط یکی از آنزیمهای القاپذیر کبدی به نام تیروزین a- کتوگلوتارات ترانسآمیناز
2- تبدیل p– هیدروکسی فنیل پیروات به هموژانتیزات: هیدروکسیلاسیون حلقه همزمان با جابجایی زنجیره جانبی. اسکوربات، احیاءکننده فیزیولوژیک این واکنش بوده و مبتلایان به اسکوربوت، محصولات نیمه اکسیده کاتابولیسم تیروزین را دفع مینمایند.
3- بازشدن حلقه آروماتیک بنزنی هموژانتیزات به مالئیل استواستات: شکست اکسیداتیو حلقه بنزنی هموژانتیزات توسط هموژانتیزات اکسیداز کبد پستانداران به مالئیل استواستات.
4- ایزومریزاسیون سیس، ترانس مالئیل استواستات و تشکیل فوماریل استواستات: ایزومریزاسیون مالئیل استواستات توسط مالئیل استواستات سیس، ترانس ایزومراز و تشکیل فوماریل استواستات.
5- هیدرولیز فوماریل استواستات و تشکیل فومارات و استواستات: هیدرولیز فوماریل استواستات و توسط فوماریل استواستات هیدرولاز و تشکیل فومارات و استواستات. استواستات میتواند تحتتأثیر β-کتوتیولاز به استیلکوآ و استات تبدیل شود.
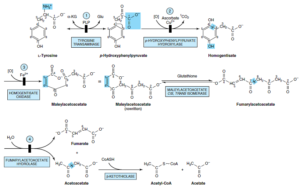
اختلالات متابولیسم تیروزین شامل موارد زیر میباشد:
1- تیروزینمی نوع I (تیروزینوز): در تیروزینمی نوع I (تیروزینوز)، احتمالاً نقص متابولیک در فوماریل استواستات هیدرولاز است. میزان تیروزین و متیونین پلاسما بالا میباشد. شیرخواران مبتلا به تیروزینوز حاد، دچار اسهال، استفراغ و بویی شبیه به کلم هستند و رشد آنها مختل میباشد. در صورت عدم درمان، نارسایی کبد ظرف 8- 6 ماه موجب مرگ آنها خواهد شد.
علائم تیروزینمی مزمن به همین صورت اما خفیفتر است و بیمار تا سن 10 سالگی عمر میکند. درمان شامل رژیم غذایی کم تیروزین و کم فنیلآلانین و در برخی موارد، کم متیونین میباشد.
2- تیروزینمی نوع II (سندروم رخنر- هانهارت): تیروزین ترانسآمیناز کبدی، محل احتمالی نقص متابولیک تیروزینمی نوع II میباشد. در این اختلال، سطح تیروزین پلاسما بالا بوده و بیماران دارای علائمی از قبیل ضایعات چشم و پوست و عقبماندگی متوسط ذهنی میباشند. در این اختلال، تیروزین تنها اسیدآمینهای است که غلظت ادراری آن بالا است. متابولیتهای ادراری شامل p- هیدروکسی فنیل پیروات، p- هیدروکسی فنیل لاکتات، p- هیدروکسی فنیل استات، N- استیل تیروزین و تیرامین میباشند.
3- تیروزینمی نوزادی: این اختلال بر اثر نقص آنزیمی p- هیدروکسی فنیل پیروات هیدروکسیلاز ایجاد میشود. سطح تیروزین و فنیلآلانین خون و نیز سطح ادراری تیروزین، – هیدروکسی فنیل استات، N- استیل تیروزین و تیرامین بالا میباشند. درمان این اختلال شامل مصرف رژیم غذایی کم پروتئین میباشد.
4- آلکاپتونوری: در این اختلال، نقص همژانتیزات اکسیداز وجود دارد. اصلیترین تظاهر بالینی آن تیره شدن ادرار متعاقب تماس با هوا میباشد. در اواخر سیر بیماری، بافت همبند به طور عمومی پیگمانته شده (آکرونوز) (Ochronosis) و شکلی از آرتریت ایجاد مینماید. آکرونوز بر اثر اکسیداسیون هموژانتیزات با پلیفنیل اکسیداز که بنزوکینون استات میسازد، ایجاد میشود که پلیمریزه شده و به ماکروملکولهای بافت همبند، اتصال مییابد. هموژانتیزات ادرار نیز با O2 موجود در هوا اکسید شده و رنگدانهای به رنگ قهوهای مایل به سیاه ایجاد مینماید.
ساخت ملانین
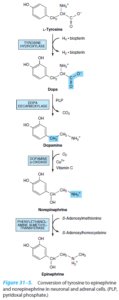
تیروزین توسط تیروزین هیدروکسیلاز (وابسته به مس) به دوپا (3 و 4- دی هیدروکسی فنیلآلانین) و سپس به دوپاکوئینون و در نهایت به ملانین تبدیل میشود.
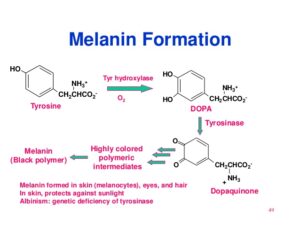
زالی (آلبینیسم):
زالی (آلبینیسم) به طیف وسیعی از سندرومهای بالینی که با هیپوملانوز ناشی از نقص ارثی ملانوسیتهای چشم و پوست همراه هستند، اطلاق میگردد. اشکال مختلف زالی چشمی پوستی، دارای کاهش پیگمنتاسیون پوست و چشم هستند که میتوان آنها را توسط مشخصات بالینی، بیوشیمیایی، فراساختمانی و ژنتیکی از یکدیگر افتراق داد.
زالهای فاقد تیروزین هیدروکسیلاز: فاقد هرگونه رنگدانه بینایی هستند. پیاز موی برداشته شده این بیماران نمیتواند تیروزین اضافه شده را به رنگدانه (پیگمان) تبدیل کند و ملانوزومهای ملانوسیتها فاقد رنگدانه هستند.
زالهای دارای تیروزین هیدروکسیلاز: دارای مقداری رنگدانه قابل مشاهده هستند و رنگ موی آنها زرد روشن متمایل به سفید میباشد. ملانوسیتهای پیاز موی آنها ممکن است ملانوزومهای مختصر رنگدانهدار داشته باشند. این ملانوزومها، تیروزین را در خارج از بدن به ملانین تبدیل میکنند.
زالی چشمی: هم به صورت اتوزومی مغلوب و هم به صورت صفت وابسته به X ایجاد میشود. ملانوسیتهای زالهای چشمی هتروزیگوت وابسته به X (نه اتووزمی مغلوب)، حاوی ملانوزومهایی درشت هستند. شبکیه زنان هتروزیگوت برای زالی چشمی وابسته به X دارای نوعی الگوی موزائیکی از توزیع رنگدانههاست که به علت غیرفعال شدن تصادفی کروموزومهای X میباشد. مشخص نیست که چه نقص متابولیکی باعث هیپوملانوز در زالی چشمی میشود.
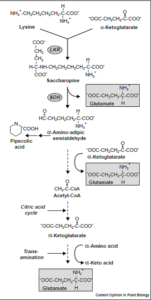
لیزین
اسکلت کربنی لیزین در پستانداران، به طور کامل به α- آمینوآدیپات و α- کتوآدیپات تبدیل میشود. ابتدا L- لیزین با α- کتوگلوتارات تشکیل یک باز شیف داده که به ساخاروپین احیا شده و سپس با دهیدروژناز دیگری، احیا میگردد. اضافه شدن آب موجب تشکیل L- گلوتامات و L- α- آمینوآدیپات γ- سمیآلدئید میشود.
α- آمینوآدیپات با ترانسآمیناسیون به α- کتوگلوتارات تبدیل میشود. احتمالاً متعاقب این واکنش، دکربوکسیلاسیون اکسیداتیو به گلوتاریل کوآ صورت میگیرد.
در مسیر تبدیل – لیزین و α- کتوگلوتارات به ساخاروپین، دو اختلال نادر متابولیک ممکن است رخ دهد:
1- هیپرلیزینمی دورهای (Periodic hyperlysinemia) همراه با هیپرآمونمی: خوردن مقادیر طبیعی پروتئین باعث شروع هیپرلیزینمی میشود، در آن صورت، میزان بالای لیزین در کبد، باعث مهار رقابتی آرژیناز کبدی گردیده و هیپرآمونمی ایجاد مینماید. مایعدرمانی و محدودسازی دریافت لیزین، موجب رفع هیپرآمونمی و تظاهرات بالینی آن میگردد. برعکس، تجویز بار لیزینی، موجب شروع حملات شدید و اغما میگردد.
2- هیپرلیزینمی ماندگار بدون هیپرآمونمی: برخی از این بیماران دارای عقبماندگی ذهنی میباشند. هیچگونه هیپرآمونمی حتی در صورت تحمیل لیزین وجود ندارد. کاتابولیتهای لیزین میتوانند در مایعات زیستی جمع شده یا جمع نشوند. ظاهراً هیپرلیزینمی ماندگار به صورت نوعی صفت اتوزومی مغلوب به ارث میرسد. علاوه بر اختلال در تبدیل لیزین و α- کتوگلوتارات به ساخاروپین، برخی از بیماران قادر به شکست ساخاروپین نیستند.
تریپتوفان
تریپتوفان تحتتأثیر تریپتوفان اکسیژناز (تریپتوفان پیرولاز)، تولید N- فرمیل کینورنین (شکستن حلقه ایندول و اضافه شدن دو اتم اکسیژن ملکولی به آن) میکند. گروه فرمیل N- فرمیل کینورنین توسط کینورنین فرمیلاز کبدی برداشته شده و تولید کینورنین مینماید. کینورنین خود میتواند در ادامه دو مسیر را در پیش بگیرد:
1- آمین خود را از طریق ترانسآمیناسیون از دست داده و تولید 2- آمینو 3- هیدروکسی بنزوئیل پیروات نموده که آن نیز با از دست دادن آب، تولید اسید کینورنیک مینماید.
2- کینورنین میتواند تولید 3- هیدروکسی کینورنین و سپس 3- هیدروکسی آنترانیلات نماید.
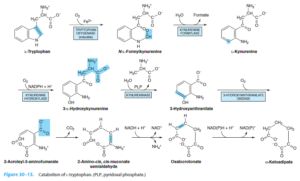
بیماری هارتناپ:
بیماری هارتناپ نوعی صفت اتوزومی مغلوب بوده که بر اثر نقص کلیوی و رودهای انتقال اسیدهای آمینه خنثی از جمله تریپتوفان ایجاد میشود. از نشانههای آن وجود همه نوع اسیدآمینه خنثی در ادرار و عموماً افزایش دفع مشتقات ایندول که از تجزیه تریپتوفان جذب نشده توسط باکتریهای روده ایجاد میشود، میباشد.
در این بیماری، اختلال جذب رودهای و بازجذب کلیوی تریپتوفان موجب کاهش ساخت نیاسین و ایجاد علائم و نشانههای شبه پلاگر میشود.
دفع گزانتورنات:
کینورنیناز یک آنزیم دارای PLP بوده که کینورنین و هیدروکسی کینورنین را به هیدروکسی آنترانیلات تبدیل مینماید. کمبود B6 موجب ناتوانی نسبی در کاتابولیسم این مشتقات کینورنینی و در نتیجه، تشکیل گزانتورنات میشود که در ادرار دفع میگردد، بنابراین، خوردن زیاد تریپتوفان در کمبود B6 موجب دفع گزانتورنات میشود.
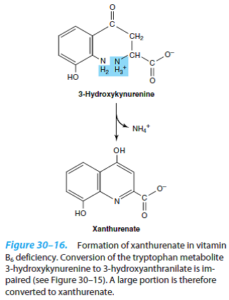
تبدیل ناکافی تریپتوفان به اسید نیکوتینیک برای ساخت نوکلئوتیدهای پیریدینی که در کمبود B6 اتفاق میافتد، میتواند به ساخت NAD+ و NADP+ لطمه بزند.
سروتونین:
تریپتوفان توسط تریپتوفان هیدروکسیلاز کبدی به 5- هیدروکسی تریپتوفان تبدیل میشود که سپس توسط دکربوکسیلاسیون به سروتونین (5- هیدروکسی تریپتامین) تبدیل میشود. سروتونین یک ترکیب منقبضکننده عروق و محرک انقباض عضلات صاف میباشد. کاتابولیسم سروتونین با دزآمیناسیون اکسیداتیو به 5- هیدروکسی ایندول استات توسط منوآمین اکسیداز (MAO) آغاز میشود که متابولیت اخیر در انسان از طریق ادرار دفع میگردد (روزانه mg 2-8).
تحریک روانی متعاقب تجویز ایپرونیازید را به قابلیت آن در مهار MAO و در نتیجه، طولانی ساختن اثر سروتونین نسبت میدهند.
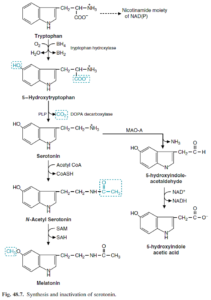
کارسینوئید بدخیم (آرژنتافینوما):
سلولهای توموری مولد سروتونین در بافت آرژنتافین حفره شکمی در کارسینوئید (آرژنتافینوما)، بیش از حد سروتونین میسازند. کاتابولیتهای سروتونینی موجود در ادرار مبتلایان به کارسینوئید شامل N- استیل سروتونین و 5- هیدروکسی ایندول استیک اسید (5-HIAA) میباشد.
از آن جایی که افزایش تبدیل تریپتوفان به سروتونین باعث کاهش ساخت اسید نیکوتینیک میشود، بیماران کارسینوئیدی ممکن است علائم پلاگر را نیز نشان دهند.
اسیدهای آمینه شاخهدار
هر 3 اسیدآمینه شاخهدار (والین، لوسین و ایزولوسین)، تحتتأثیر یک ترانسآمیناز واحد به α- کتواسید تبدیل میشوند، سپس این α- کتواسیدهای شاخهدار توسط یک کمپلکس چند آنزیمی میتوکندریایی موسوم به دهیدروژناز α- کتواسیدهای شاخهدار، دکربوکسیله شده و تولید تیواسترهای شاخهدار را مینمایند. این تیواسترهای شاخهدار نیز به نوبه خود دهیدروژنه میشوند.
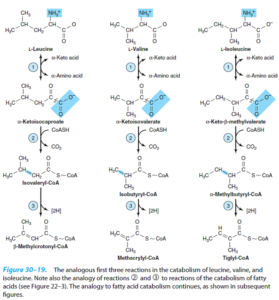
اختلالات متابولیک اسیدهای آمینه شاخهدار شامل موارد زیر میباشند:
1- بیماری شربت افرا (کتونوری شاخهدار): در این بیماری اتوزومی مغلوب، فقدان یا کاهش فعالیت آنزیم α- کتواسید دکربوکسیلاز وجود دارد. ادرار این بیماران دارای بوی شربت افرا یا قند سوخته میباشد. میزان والین، لوسین، ایزولوسین و α- کتواسیدهای آنها در خون و ادرار افزایش مییابد. بیماری تا انتهای هفته اول تولد، آشکار میشود. نوزاد شیرخوار به سختی شیر میخورد و ممکن است استفراغ کند یا خوابآلود باشد. تشخیص پیش از 1 هفتگی فقط با تحلیل آنزیمی یا ژنتیکی امکانپذیر است. کودکانی که زنده میمانند، دچار آسیب وسیع مغزی شده و در صورت عدم درمان، معمولاً پیش از 1 سالگی، میمیرند.
درمان آن شامل جایگزینی پروتئین غذا با مخلوطی از اسیدهای آمینهی فاقد والین، لوسین و ایزولوسین میباشد. پس از کنترل سطح پلاسمایی این اسیدهای آمینه، میتوان آنها را به مقدار موردنیاز به بدن رساند.
2- کتونوری شاخهدار متناوب: این اختلال احتمالاً به دلیل تغییر ساختمانی خفیفتری در α- کتواسید دکربوکسیلاز میباشد. افراد مبتلا قابلیت کاتابولیسم والین، لوسین و ایزولوسین را اگرچه به شکل مختلشده، دارند و از این رو، علائم بیماری دیرتر ظاهر میشود. پیشآگهی آن هم با رژیم درمانی، بهتر است.
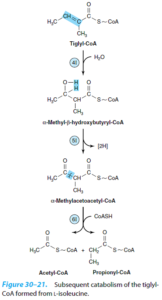
3- ایزووالریک اسیدمی: نقصی در کاتابولیسم لوسین میباشد. آنزیم ایزووالریل کوآ دهیدروژناز نقص دارد. در این بیماران، ایزووالریل کوآ تجمع یافته و به ایزووالرات هیدرولیز میگردد که از طریق ادرار و عرق دفع میشود. علائم آن با خوردن پروتئین زیاد آغاز میشود و شامل «بوی پنیری» تنفس و مایعات بدن، استفراغ، اسیدوز و اغما میباشد.
4- متیل مالونیک اسیدوری: در ادامه کاتابولیسم متاکریلیل کوآ حاصل از والین و طی چند واکنش، متیل مالونیل کوآ ایجاد میشود که با کمک ویتامین B12 به سوکسینیل کوآ ایزومریزه میشود. در کمبود B12 میتواند متیل مالونیک اسیدوری ایجاد شود. 2 شکل از این بیماری شناخته شده که یکی از آنها به دوزهای فارماکولوژیک و دیگری تنها به دوزهای فوقالعاده زیاد (روزانه 1 گرم) ویتامین B12 پاسخ میدهد.
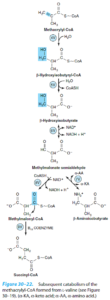
5- پروپیونیک اسیدمی: در ادامه کاتابولیسم تیگلیل کوآ حاصل از ایزولوسین و طی چند واکنش، پروپیونیل کوآ حاصل میشود. کمبود پروپیونیل کوآ کربوکسیلاز با میزان بالای پروپیونات سرمی مشخص میگردد. درمان آن شامل رژیم غذایی کم پروتئین و اقداماتی برای مقابله با اسیدوز متابولیک میباشد.
ساخت گابا (GABA) (γ– آمینو بوتیرات)
L-گلوتامات توسط L-گلوتامات دکربوکسیلاز (وابسته به PLP) که در بافتهای دستگاه مرکزی اعصاب و عمدتاً ماده خاکستری وجود دارد، به GABA تبدیل میشود.
پوترسین نیز از 2 طریق به GABA تبدیل میشود:
1- از طریق دزآمیناسیون آن با دیآمین اکسیداز
2- از طریق واسطههای N- استیله
کاتابولیسم GABA:
GABA با ترانسآمیناسیون توسط γ- آمینوبوتیرات ترانسفراز به سوکسینات سمیآلدئید تبدیل میشود، سپس سوکسینات سمیآلدئید ممکن است به L- لاکتات دهیدروژناز احیاء شود یا به سوکسینات اکسید گردد و از طریق چرخه اسید سیتریک به CO2 و H2O تبدیل شود.
نقص گابا ترانسآمیناز:
نقص گابا ترانسآمیناز یک اختلال فوقالعاده نادر ژنتیکی در متابولیسم گابا است. این آنزیم در کاتابولیسم گابا پس از آزادی پسسیناپسی آن در بافت مغز شرکت دارد. این اختلال نوعی ناهنجاری اتوزوم مغلوب است که با اختلال رشد و تشنج خود را نشان میدهد.
بادرنجبویه (Melissa officinalis) دارای بازدارنده فعالیت GABA ترانسآمیناز بوده که این خاصیت بادرنجبویه اثر ضد اضطرابی آن را توضیح میدهد. عصاره بادرنجبویه میتواند باعث بهبود خلق و خوی و عملکرد مغزی شود. رزمارینیک اسید نیز دارای بازدارنده فعالیت GABA ترانسآمیناز میباشد.
از بازدارندههای GABA ترانسآمیناز به داروهای والپروئیک اسید، ویگاباترین، L- سیکلوسرین و … میتوان اشاره نمود که به داروهای ضد تشنج موسوم هستند.
هیپر β– آلانینمی: در پستانداران، β- آلانین به صورت کاتابولیتی از سیتوزین، کارنوزین و آنسرین ایجاد میشود. در بافتهای پستانداران، β- آلانین با ترانسآمیناسیون به مالونات سمیآلدئید تبدیل میشود که از اکسیداسیون آن، استات و در نهایت CO2 ایجاد میشود. در اختلال متابولیکی نادر هیپر β- آلانینمی، میزان β- آلانین، تورین و β- آمینو ایزوبوتیرات مایعات و بافتهای بدن افزایش مییابد.
فئوکروموسیتوما
فئوکروموسیتوما توموری است که از قسمت مرکزی آدرنال منشأ میگیرد و سن شیوع آن اغلب دهه چهارم و پنجم زندگی میباشد. این تومور ممکن است در نواحی خارج آدرنال (گردن، شکم و …) هم دیده شود که به پاراگانگلیوما موسوم است.
علائم آن شامل تریاد کلاسیک تپش قلب، تعریق و سردرد است. در کنار این علائم، میتواند علائم دیگری نیز مانند تهوع، استفراغ، درد شکم و فلاشینگ وجود داشته باشد که غیراختصاصی هستند. از دیگر علائم آن میتوان به فشار خون بالا اشاره نمود.
از راههای تشخیصی آن میتوان به اندازهگیری وانیلیل ماندلیک اسید (VMA) در ادرار 24 ساعته اشاره نمود.
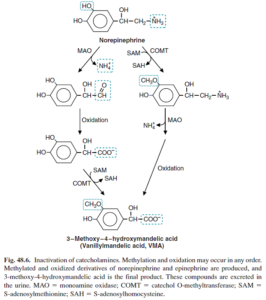
وانيليل ماندليك اسيد (VMA) (Vanillylmandelic acid):
وانيليل ماندليك اسيد (VMA)، متابوليت اصلي ناشي از متابوليسم كاتكولآمينها (اپينفرين و نوراپينفرين) ميباشد. در هنگام افزايش كاتكولآمينها، ميزان توليد VMA نيز افزايش مييابد. براي اين آزمايش جمعآوري ادرار 24 ساعته لازم ميباشد. استرس، هيپوگليسمي، هيپرتيروئيديسم، ورزش، استعمال دخانيات و برخي داروها ميتوانند منجر به افزايش توليد كاتكولآمينها و در نتيجه افزايش VMA شوند. اسكن راديواكتيو ميتواند به مدت يك هفته در اين آزمايش تداخل نمايد.
منابع:
1-Tietz Textbook of Clinical Chemistry and Molecular Diadnosis. 2006; 4th Edition.
2- Henrys Clinical Diagnosis and Management by Laboratory Methods. 2007; 21st Edition.
3 – Wendy Aineson and Jean Brickell. Clinical Chemistry; A Laboratory Perspective. 2007; 1st Edition.
4- Arneson W, Brickell J. Clinical chemistry; a laboratory perspective. 2007.
5- Van Leeuwen AM, Kranpitz TR and Smith L. Laboratory and diagnostic tests with nursing implications. 2006; 2nd Edition.
6-Rodwell VW, Bender DA, Botham KM, Kennelly PJ and Weil PA. Harpers illustrated biochemistry. 2015; 30th Edition.
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام