اختلالات کبدی و بررسیهای آزمایشگاهی آنها
مراد رستمي: کارشناس ارشد بیوشیمی بالینی، دانشگاه علوم پزشكي جندي شاپور اهواز
معصومه جرفی: کارشناس ارشد میکروب شناسی، دانشگاه علوم پزشكي جندي شاپور اهواز
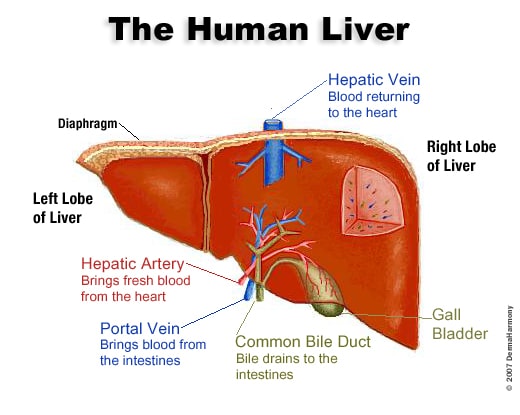
ساختمان کبد
کبد، بزرگترین و پیچیدهترین اندام دستگاه گوارش است که شامل سیستمهای زیر میباشد:
1- سیستم بیوشیمیایی هپاتوسیتی
2- سیستم کبدی صفراوی (هپاتوبیلیاری)
3- سیستم رتیکولواندوتلیال
عملکرد کبد
سیستم بیوشیمیایی هپاتوسیتی:
این مسیر، مسئول عمده فعالیتهای متابولیکی بدن شامل سنتز پروتئینها، متابولیسم هوازی و بیهوازی گلوکز و سایر قندها، سنتز و تجزیه گلیکوژن، متابولیسم اسیدهای آمینه و اسید نوکلئیک، تبدیل اسید آمینه و اسید دیکربوکسیلیک به یکدیگر از طریق ترانسآمینازها (آمینوترانسفرازها)، سنتز و متابولیسم لیپوپروتئین، متابولیسم گزنوبیوتیک، ذخیره آهن و ویتامینهایی از قبیل A ،D و B12 و سنتز هورمونهایی از قبیل آنژیوتانسینوژن، فاکتور رشد شبه انسولین 1 (IGF-1) و تری یدو تیرونین (T3) میباشد. این سیستم همچنین محل کلیرانس اکثر هورمونها از قبیل انسولین، هورمون پاراتیروئید (PTH)، استروژن و کورتیزول میباشد. کبد تنها محل متابولیسم امونیاک به اوره میباشد. کل آلبومین بدن و نیز تمامی فاکتورهای انعقادی بجز فاکتور فونویلبراند که در مگاکاریوسیتها و سلولهای اندوتلیال ساخته میشود، همگی در کبد سنتز میشوند.
سیستم کبدی صفراوی (هپاتوبیلیاری):
این سیستم با متابولیسم بیلیروبین (شامل انتقال بیلیروبین به درون هپاتوسیتها، کنژوگه شدن آن با اسید گلوکورونیک و ترشح آن به درون مجاری صفراوی و سیستم کبدی- روده ای) در ارتباط است.
سیستم رتیکولواندوتلیال:
این سیستم شامل سلولهای کوپفر (نوعی ماکروفاژ) میباشد که در دفاع علیه باکتریها و برداشت کمپلکسهای آنتیژن- آنتیبادی از گردش خون نقش دارد. این سلولها همچنین محل تجزیه هموگلوبین حاصل از گلبولهای قرمز مرده هستند.
اعمال متابولیک کبد
آمونیاک (Ammonia)
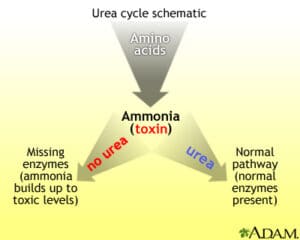
آمونیاک به طور عمده از متابولیسم اسیدهای آمینه و اسیدهای نوکلئیک تشکیل میشود. مقداری آمونیاک نیز از طریق واکنشهای متابولیکی (مانند اثر گلوتامیناز بر گلوتامین و تشکیل اسید گلوتامیک و آمونیاک)، توسط توبولهای کلیوی تولید میشود. آمونیاک در کلیهها به عنوان یک بافر کلیوی مهم به شمار میرود.

آمونیاک از طریق چرخه اوره در کبد به اوره که یک ترکیب غیر سمی است، تبدیل شده و دفع میگردد.
| Location | Abb. | Enzyme | Disorder | Measurements |
| Mitochondria | NAGS | N-Acetylglutamate synthetase | N-Acetylglutamate synthase deficiency | +Ammonia |
| Mitochondria | CPS1 | Carbamoyl phosphate synthetase I | Carbamoyl phosphate synthetase I deficiency | +Ammonia |
| Mitochondria | OTC | Ornithine transcarbamylase | Ornithine transcarbamylase deficiency | +Ornithine, +Uracil, +Orotic acid |
| Cytosol | ASS | Argininosuccinic acid synthetase | “AS deficiency” or citrullinemia | +Citrulline |
| Cytosol | ASL | Argininosuccinase acid lyase | “AL deficiency” or argininosuccinic aciduria (ASA) | +Citrulline, +Argininosuccinic acid |
| Cytosol | ARG | Arginase | “Arginase deficiency” or argininemia | +Arginine |
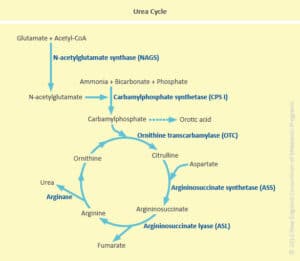
چنانچه هر گونه اختلالی در آنزیمهای چرخه اوره رخ دهد، منجر به تجمع و افزایش سطح آمونیاک در گردش خون و مایع مغزی- نخاعی خواهد شد. آزمایش آمونیاک میتواند برای تعیین این که آیا نارسایی کبدی، مسئول ایجاد علائم گیجی، خواب آلودگی، کوما و یا لرزش دستها در فرد میباشد و یا نه نیز درخواست شود. همچنین از این آزمایش ممکن است به منظور ارزیابی مفید بودن داروها و تاثیر درمان در بیماری های کبدی از قبیل سیروز نیز استفاده شود.
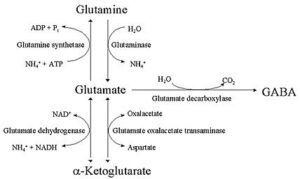
علت ایجاد سمیت بر سیستم عصبی مرکزی در هنگام افزایش آمونیاک خون، به واسطه کاهش غلظت گاماآمینوبوتیریک اسید (GABA) رخ میدهد. کاهش GABA که یک نروترانسمیتر مهم در CNS میباشد، از طریق واکنش با اسید گلوتامیک برای تشکیل گلوتامین میباشد که از طریق واکنش معکوس کاتالیز شده به وسیله گلوتامیناز صورت میگیرد. بنابراین، اسید گلوتامیک کاهش یافته و GABA نیز که از دکربوکسیلاسیون اسید گلوتامیک به دست میآید، مرتباً کاهش مییابد.
روش اندازهگیری: گلوتامات دهیدروژناز، واکنش آمونیاک با آلفاکتوگلوتارات برای تشکیل گلوتامات همراه با اکسیداسیون NADPH به NADP (کاهش جذب در 340 نانومتر) را کاتالیز میکند.
نمونه مورد نیاز: نمونه خون شریانی (گاهی وریدی) غیر همولیز که روی ضد انعقاد EDTA گرفته میشود. نمونه باید بلافاصله روی ظرف یخ قرار گیرد. برای انجام این آزمایش، بایستی فرد حداقل به مدت 8 ساعت ناشتا باشد و از انجام فعالیتهای شدید و استعمال دخانیات قبل از انجام آزمایش، خودداری نماید.
نحوه گزارش: مقدار طبیعی آن در نوزادان، 150- 90 میکروگرم در دسیلیتر (107- 64 میکرومول در لیتر)، در کودکان، 80- 40 میکروگرم در دسیلیتر (57- 28 میکرومول در لیتر) و در بالغین، 45- 15 میکروگرم در دسیلیتر (32- 11 میکرومول در لیتر) میباشد.
لیپیدها
کبد، اندامی حیاتی در سنتز و تبدیلات بینابینی لیپوپروتئینها میباشد؛ بنابراین، اختلالات کبدی میتوانند موجب اختلال در متابولیسم لیپوپروتئینها شوند. در آسیبهای شدید کبدی نظیر سیروز، کاهش HDL به ویژه جزء HDL3 و سایر اختلالات توزیع لیپوپروتئین، به ویژه به علت نقصهای لسیتین کلسترول آسیل ترانسفراز (LCAT) و لیپوپروتئین لیپاز (LPL) که باعث هیپرتریگلیسریدمی (مقادیر تریگلیسرید در محدوده 500- 250 میلیگرم در دسیلیتر) میشوند، میتوانند رخ دهند.

در آسیب کبدی القاء شده توسط الکل، الکل باعث القای افزایش بیان پروتئین apo-A1 میشود. بنابراین اگر خوردن الکل ادامه یابد، ممکن است HDL و به ویژه HDL3 افزایش یابد.
در سیروز، پروتئین apo-A1 کاهش مییابد که برای تشخیص آن میتوان از شاخص PGA (PGA Index) استفاده نمود. شاخص PGA شامل مجموع نمرات حاصل از نتایج 3 آزمایش زمان پروترومبین (PT)، گاما گلوتامیل ترانسفراز (GGT) و آپولیپوپروتئین A1 (Apo A-I) میباشد. برای هر کدام از این آنالیتها، محدوده نمرات صفر تا 4 در نظر گرفته شده است که متناسب با افزایش شدت آسیب کبدی ایجاد شده و افزایش شدت بیماری، نمره هر آنالیت نیز همان طور که در جدول زیر نشان شده است، افزایش مییابد.
برای Apo A-I، افزایش شدت بیماری، مرتبط با کاهش غلظت این پروتئین در سرم میباشد. در نهایت، مجموع 3 نمره حاصل از آزمایشهای PT، GGT و Apo A-I با هم جمع شده و بدین ترتیب، شاخصی از میزان فیبروز کبدی و شدت سیروز، به دست میدهد. نشان داده شده است که در بیماران با بیماری کبدی ناشی از مصرف الکل، شاخص PGA کمتر از 6 میتواند با احتمال 90%، وجود سیروز را رد نماید. در یک مطالعه دیگر نیز نشان داده شده است که شاخص PGA دارای حساسیت و اختصاصیت به ترتیب، 91% و 81% میباشد.
جدول ارتباط بین نتایج حاصل از آزمایشهای PT، GGT و Apo A-I با نمرات هر کدام از آنها
| نمره | نتیجه آزمایش | نام آزمایش |
| 0 | 80 ≤ | درصد فعالیت زمان پروترومبین (PT) |
| 1 | 79- 70 | |
| 2 | 69- 60 | |
| 3 | 59- 50 | |
| 4 | 50 > | |
| 0 | 20 > | گاما گلوتامیل ترانسفراز (GGT) |
| 1 | 20-49 | |
| 2 | 99- 50 | |
| 3 | 199- 100 | |
| 4 | 200 ≤ | |
| 0 | 200 ≤ | آپولیپوپروتئین A1 (Apo A-I) |
| 1 | 199- 175 | |
| 2 | 174- 150 | |
| 3 | 149- 125 | |
| 4 | 125 > |
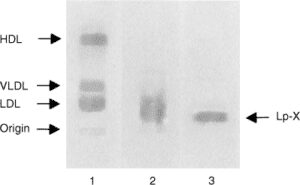
در کلستاز، برگشت محتویات صفراوی به داخل خون موجب افزایش ساخت لیپوپروتئین X (LPX) میشود که منجر به افزایش لیپیدهای صفراوی میشود. چون LPX مقادیر بالایی از کلسترول غیر استریفیه را در خون حمل میکند، مقادیر کلسترول در سرم به طور بارزی میتواند افزایش یابد.
LPX، یک لیپوپروتئین غیر طبیعی یافت شده در بیماران با انسداد مجاری صفراوی و در بیماران با نقص فامیلی LCAT میباشد. بیش از 90% وزن آن از لیپیدها (اکثرا فسفولیپیدها، کلسترول استری نشده و مقدار بسیار کمی کلسترول استری شده) و کمتر از 10% وزن آن از پروتئینها (عمدتا آپو C و میزان کمتری آلبومین) تشکیل شده است.
نمکهای صفراوی
نمکهای صفراوی، محصولات متابولیسم کلسترول بوده و در تسهیل جذب چربی از رودهها نقش دارند. نمکهای صفراوی در کیسه صفرا ذخیره شده و پس از مصرف غذا، از طریق انقباض کیسه صفرا توسط کوله سیستوکینین، به درون روده آزاد میشوند. نمکهای صفراوی معمولاً در تشخیص اختلالات عملکردی کبد به کار نمیروند، اما در ایجاد مقادیر قابلتوجهی صفرا برای دفع بیلیروبین حائز اهمیت هستند و از این رو میتوانند در تشخیص کلستاز به کار روند. در انسداد مجاری صفراوی، نمکهای صفراوی در سرم تجمع یافته و باعث خارش شدید میشوند. نمکهای صفراوی اولیه (کولات و کنوداکسی کولات) در کبد تولید شده و سپس به درون سیستمهای صفراوی و کبدی- رودهای (انتروهپاتیک) ترشح میشوند.
در سیستم رودهای، توسط باکتریها متابولیزه شده و در اثر واکنش آلفا دهیدروکسیلاسیون، تولید نمکهای صفراوی ثانویه (لیتوکولات و داکسیکولات) را مینمایند.
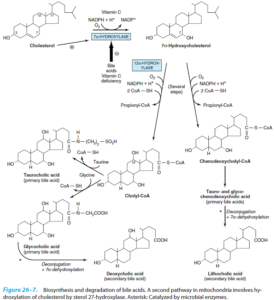
نمکهای صفراوی در سیستم میکروزومی توسط گلیسین و تورین، کنژوگه و همچنین سولفاته میشوند. کنژوگاسیون نمکهای صفراوی با تورین و سولفاتها، با شدت کلستاز افزایش یافته و در شرایطی باعث ایجاد انسداد برونده صفراوی میشوند. بازگردش نمکهای صفراوی به کبد، از طریق بازجذب آنها از لومن روده اتفاق میافتد. داکسیکولات تقریباً به طور کامل و کنوداکسیکولات در حدود 75% بازجذب میشود.
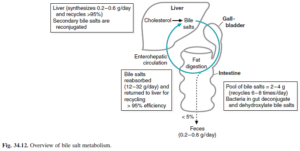
در سیروز، کاهش نامتناسبی در اسید کولیک و نسبت کاهش یافته نمکهای صفراوی اولیه به ثانویه وجود دارد. در کلستاز، نمکهای صفراوی ثانویه تشکیل نمیشود، بنابراین نسبت نمکهای صفراوی اولیه به ثانویه، به طور بارزی افزایش مییابد.
کلیرانس کلیوی نمکهای صفراوی در بیماران عادی ناچیز است، اما در کلستاز، دفع کلیوی نمکهای صفراوی عمدتاً به فرم سولفاته و گلوکورونیده افزایش مییابد. نمکهای صفراوی ناشتایی، هنگامی که طبیعی هستند، وجود بیماری پارانشیمال کبدی را رد مینمایند. تولید معیوب نمکهای صفراوی (که به حلالیت محتوای صفرا کمک میکنند) در کبد، ممکن است سبب تشکیل سنگهای بیلیروبینی یا کلسترولی و انسداد مجاری صفراوی پس کبدی شوند.
آنالیز نمکهای صفراوی بر روی سرم بیماران باید در حالت ناشتایی صورت گیرد؛ زیرا خوردن غذا موجب افزایش میزان اسیدهای صفرائی میگردد. نمکهای صفراوی میتوانند توسط تکنیکهای زیادی از جمله HPLC اندازهگیری شوند.
متابولیسم دارو
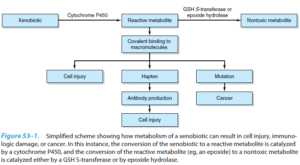
اکثر گزنوبیوتیکها از قبیل داروها، عمدتاً در میکروزوم سلولهای کبدی و توسط چند سری از واکنشها که اکثراً وابسته به سیتوکروم P450 هستند، متابولیزه میشوند. تبدیل گزنوبیوتیکها به متابولیتها از طریق این سیتم اغلب شامل دو فاز است؛ واکنشهای فاز I شامل اکسیداسیون/ هیدروکسیلاسیون بوده و واکنشهای فاز II مسئول کنژوگهسازی متابولیتها و یا ترکیبات اصلی با برخی ترکیبات مانند اسید گلوکورونیک، گلیسین، تورین و سولفات میباشد. در بیماریهای خیلی شدید کبدی (که شامل آسیب میکروزومی میباشد)، توانایی کبد برای متابولیزه کردن گزنوبیوتیکها دچار اختلال میشود، از این رو، توانایی سلولهای کبدی برای متابولیزه کردن دارو میتواند در ارزیابی آسیب کبدی به کار رود. برای این کار دوز مشخصی از داروی نشاندار شده با 13C به بیمار تزریق شده و پس از گذشت زمان خاصی، 13CO2 بازدمی تنفس بیمار اندازهگیری میشود.
تستهای تنفسی بر اساس مرحله محدودکننده سرعت شامل دو گروه میباشند:
1- گروه اول شامل داروهایی مانند آمینوپیرین، کافئین و دیازپام بوده که صرفاً وابسته به فعالیت آنزیمی سیتوکرومهای P450 متابولیزه میشوند.
2- گروه دوم شامل داروهایی مانند متاستین، فناستین و اریترومایسین بوده که متابولیسم آنها وابسته به سرعت جریان خون میباشد.
این تستها در تخمین وسعت ضایعه کبدی در یک بیماری مشخص کبدی مفید هستند. برخی عوامل مداخلهگر، ممکن است تفسیر این تستها را با مشکل مواجه سازند. از جمله این عوامل مداخلهگر میتوان به موارد زیر اشاره نمود:
1- دمتیلاسیون آمینوپیرین (گروه متیل به CO2 تبدیل میشود) وابسته به ویتامین B12 بوده و در نقص این ویتامین، 13CO2 خارج شده، کمتر از حد طبیعی خواهد بود.
2- میزان متابولیسم کافئین عموماً با افزایش سن کاهش مییابد.
3- میزان متابولیسم کافئین در اثر مصرف سیگار، افزایش مییابد.
کبد و سنتز پروتئینها
کبد محل سنتز اکثر پروتئینهای پلاسما (به استثنای ایمونوگلبولینها و فاکتور فونویلبراند) میباشد. بیش از 90% پروتئینها و 100% سنتز آلبومین در کبد اتفاق میافتد، بنابراین تخریب وسیع بافت کبدی، باعث کاهش مقادیر پروتئین تام و آلبومین سرم خواهد شد. در سیروز، علاوه بر تخریب هپاتوسیتها، فشار زیاد پورتال نیز (که تحویل اسیدهای آمینه به کبد را کاهش میدهد) عامل کاهش تولید پروتئین میباشد. البته عوامل اصلی دیگری مانند بیماری کلیوی، سوء تغذیه، انتروپاتیهای با اتلاف پروتئین و به میزان کمتری، بیماریهای التهابی مزمن نیز در کاهش پروتئین تام و آلبومین سرم نقش دارند، بنابراین در ارزیابی وضعیت عملکردی کبد، باید این عوامل را نیز موردتوجه قرار داد.
اندازهگیری: تعیین میزان پروتئین سرم معمولاً بر اساس روش بیوره میباشد که در آن گروه های C=O موجود در اسکلت پپتیدی پروتئینها با مس، موجب ایجاد کمپلکسهای رنگی میشود که دارای جذب نوری در طول موج 540 نانومتر هستند. برخی روشها نیز از اتصالات رنگی پروتئینها با کوماسیبلو بهره میبرند. آلبومین نیز از طریق تشکیل کمپلکسهای رنگی با بروموکرزول اندازهگیری میشود. محدوده مرجع برای مقادیر سرمی پروتئین تام، 7/8- 6 و برای آلبومین، 5- 3 گرم در دسیلیتر است. در حالت عادی، حداقل 60% پروتئین تام را باید آلبومین تشکیل دهد.
الکتروفورز پروتئینهای سرم
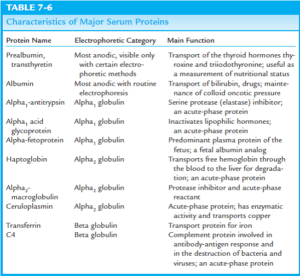
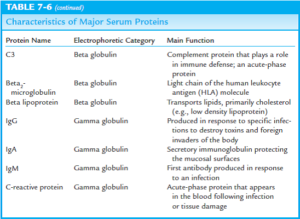
پروتئين توتال از آلبومين و گلبولينها تشكيل شده است. گلبولينها به صورت اوليه داراي 3 نوع بوده كه شامل گلبولينهاي آلفا، بتا و گاما ميباشند. آلفاگلبولينها در كبد ساخته شده و شامل آلفا- 1 گلبولينها (شامل آلفا–1- آنتيتريپسين، آلفا فيتوپروتئين و گلبولين متصل شونده به تيروكسين) و آلفا-2 گلبولينها (شامل هاپتوگلوبين، سرولوپلاسمين، HDL و ماكروگلبولين) ميباشند. بتاگلبولينها نيز در كبد ساخته شده و شامل ترانسفرين، پلاسمينوژن، LDL و پروتئينهاي كمپلمان ميباشند. گاماگلبولينها كه ايمونوگلبولينها نيز ناميده ميشوند، توسط لنفوسيتهاي Bدر پاسخ به تحريك توسط آنتيژنها ساخته شده و شامل آنتيباديهاي IgA، IgD، IgE، IgG و IgM ميباشند.
الكتروفورز پروتئينهاي سرم يك روش شايع در اندازهگيري آلبومين و هر كدام از انواع گلبولينها ميباشد. از اين آزمايش در شناسايي بيماران داراي مولتيپلميلوما و ساير اختلالات پروتئين سرمي، شرايط التهابي، بيماريهاي اتوايميون، عفونت و شرايطي كه منجر به از دست دادن پروتئين ميشوند، استفاده ميشود. آزمايش الكتروفورز پروتئينهاي سرم همچنين به منظور پيگيري ساير نتايج غيرطبيعي آزمايشها از قبيل پروتئين توتال، آلبومين، پروتئين ادرار، كاهش سطح كلسيم و كاهش تعداد گلبولهاي سفيد و (يا) گلبولهاي قرمز مورد استفاده قرار ميگيرد. اين آزمايش ميتواند به منظور مشخص كردن پيشرفت بيماري و يا پاسخ بيماري به درمان نيز مورد استفاده واقع شود.
الكتروفورز به جداسازي پروتئينها بر اساس ويژگيهاي فيزيكي آنها از يكديگر ميپردازد. نمونه سرم در يك محيط مخصوص قرار گرفته و پس از برقراري جريان الكتريكي، پروتئينها بر اساس ميزان بار، اندازه ملكولي و شكل از يكديگر جدا ميشوند.
در حركت از قطب منفي به سمت قطب مثبت، آلبومين داراي بيشترين سرعت بوده و به دنبال آن، آلفا–1- گلبولينها، آلفا– 2-گلبولينها، بتاگلبولينها و گاماگلبولينها قرار ميگيرند، سپس از روي الگوي ميزان پروتئينهاي مختلف در الكتروفورز به تشخيص وضعيت مربوطه ميپردازند.
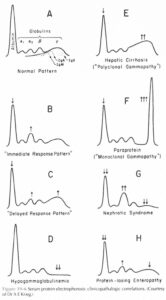
افزايش ارتفاع قله مربوطه به گاماگلبولينها به صورت نوكتيز، نشاندهنده مونوكلونال گاموپاتي بوده كه در ارتباط با شرايط بدخيم از قبيل مولتيپل ميلوما، ماكروگلبولينمي والدنشتروم (Waldenstrom’s macroglobulinemia)، سرطان خون، بيماري زنجيره سنگين و ميلوئيدوز ميباشد. پليكلونال گاموپاتي (افزايش سطح مربوط به گاماگلبولينها به صورت يك باند پهن) ميتواند در نتيجه هرگونه پروسه واكنشي و يا التهابي باشد. در هنگام وجود مونوكلونال گاموپاتي، بايد ابتدا مولتيپلميلوما از ساير علل ايجادكننده آن افتراق داده شود. اين كار ميتواند از طريق الكتروفورز ايمونوفيكساسيون (IFE) انجام شود. از IFE معمولاً متعاقب حضور باندهاي پروتئين غيرطبيعي كه ممكن است ايمونوگلبولين باشند، استفاده مي شود. افزايش کاذب سطح ايمونوگلبولينها ميتواند در ايمونيزاسيون در طي 6 ماه گذشته در فرد مشاهده شود.
مقدار طبیعی پروتئين توتال 8-6 گرم در دسيليتر ميباشد. مقدار طبیعی آلبومين 5/5-3/3 (74- 58 درصد)، آلفا-1 گلبولين 0/4- 0/1 (3/5- 2 درصد)، آلفا-2- گلبولين 1- 0/5(10/6- 5/4 درصد)، بتاگلبولين 1/2- 0/7(14- 7 درصد) و گاماگلبولين 1/6- 0/8 (18- 8 درصد) ميليگرم در دسيليتر ميباشد.
الکتروفورز پروتئینهای سرم و اندازهگیری کمی ایمونوگلبولینها ممکن است تغییرات اختصاصی در بیماری کبدی را آشکار سازد، به عنوان مثال عموماً در سیروز، آلبومین و نیز باندهای آلفا یک، آلفا دو و بتا (اساساً ترانسفرین) به طور بارزی کاهش مییابند. با این وجود، اغلب یک افزایش پلیکلونال ایمونوگلبولینها وجود دارد که الگوی ممزوج شدن باندهای بتا و گاما را ایجاد میکند. در هپاتیت اتوایمیون، کاهش آلبومین توأم با افزایش قابلتوجهی از IgG پلیکلونال مشاهده میشود.
آلبومین
آلبومین، پروتئین اصلی تولید شده توسط کبد بوده که به میزان 120 میلیگرم به ازای هر کیلو وزن بدن در روز تولید میشود. میزان سنتز آن تحت فشار انکوتیکی پایین میتواند تقریباً دو برابر شود. کاهش آلبومین یکی از نشانههای اصلی پیشآگهی در بیماران مبتلا به سیروز میباشد. مقادیر پایین آلبومین سرمی به علت بیماری کبدی تقریباً همیشه در اثر تخریب بافت کبدی ایجاد میشود که به طور اولیه در سیروز و ثانویه به مسمومیت الکلی میباشد. کاهش آلبومین با افتی در پروتئین تام سرمی همراه است. آلبومین از نظر اسمزی، یک جزء فعال داخل عروقی بوده و کاهش آن (هیپوآلبومینمی)، اغلب باعث ایجاد ادم میشود. در سیروز، که افزایش مقاومت به جریان خون در سینوزوئیدها باعث هیپرتانسیون پورتال میشود، افزایش فشار هیدروستاتیک در سیستم پورتال و کاهش فشار اسمزی معمولاً باعث ایجاد آسیت میشود که یک یافته شایع در سیروز میباشد.
آلفا-1- آنتی تریپسین (Alpha-1-Antitrypsin; AAT)
آلفا-1- آنتیتریپسین (AAT)، فراوانترین جزء آلفا-1- گلبولین و مهمترین مهارکننده پروتئازی در سرم میباشد. AAT علاوه بر تریپسین، موجب مهار سایر سرین پروتئازها نظیر الاستاز نیز میشود. طی التهاب حاد، میزان AAT میتواند چندین برابر افزایش یابد. از این رو، این پروتئین جزء پروتئینهای فاز حاد طبقهبندی شده و همانند دیگر پروتئینهای فاز حاد توسط سلولهای کبدی ساخته میشود. نقش اصلی AAT، تخریب الاستاز میباشد. الاستاز آنزیمی است که طی التهاب از سلولهای التهابی، از جمله نوتروفیلها، آزاد شده و باعث تجزیه الاستین میگردد. لازم به ذکر است که الاستین از پروتئینهای ساختاری بافت ریه بوده و عامل الاستیسیته این بافت میباشد. تخریب این پروتئین تحتتأثیر الاستاز منجر به آمفیزم ریوی و بیماریهای ریوی انسدادی مزمن COPD میگردد، لذا نقش اصلی آلفا یک آنتیتریپسین محافظت از بافت ریه در مقابل اثر تخریبی الاستاز میباشد.
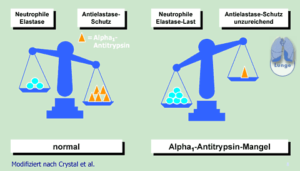
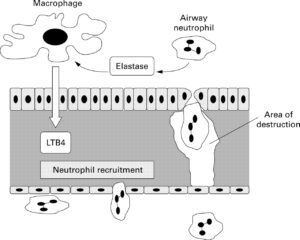
AAT توسط ژن Pi (مهارکننده پروتئاز) بر روی کروموزوم 14 کد میشود. واریانتهای ژنتیکی متعددی در نتیجه جهشهای نقطهای به وجود آمدهاندکه منجر به جایگزینی اسیدآمینهای میگردند. شایعترین واریانت، M مرتبط با مقدار AAT سرمی طبیعی میباشد. واریانتهای S و Z مانع از گلیکوزیلاسیون طبیعی پروتئین شده و منجر به تجمع AAT در داخل هپاتوسیتها و کاهش مقادیر پلاسمایی AAT میگردند. ژنوتیپهای PiZZ، PiSS، PiSZ، PiMZ و PiMS همگی به استثنای ژنوتیپ نادر نول (Pi–)، دارای فعالیت آنتیپروتئازی قابلاندازهگیری میباشند. اگر فعالیت آنتیپروتئازی فنوتیپ MM به عنوان مرجع در نظر گرفته شود، آنگاه فعالیت فنوتیپ ZZ 15%، SS 60%، MZ 5/57% و MS 80% میباشد.
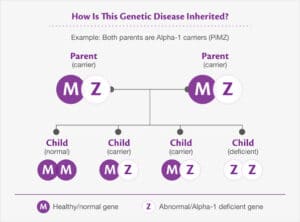
بزرگسالان دارای فنوتیپ PiZZ،مستعدترین اشخاص برای ایجاد آمفیزم نسبتاً زودرس در زندگی بوده که در نتیجه فعالیت تریپسین مهار نشده بر روی الاستین دیواره آلوئولی به وجود میآید. از آن جایی که AAT، یک واکنشگر فاز حاد میباشد، مقادیر سرمی آن در فرم هتروزیگوت MZ میتواند طبیعی باشد.
روشهای بررسی آلفا یک آنتیتریپسین:
1- اندازهگیری غلظت آلفا-1- آنتیتریپسین درسرم به روش نفلومتری
2- تعیین غلظت آلفا-1- آنتیتریپسین با روش ایمونولوژیکی
3- تعیین درصد باند آلفا-1- گلوبولین بوسیله الکتروفوزاستات سلولز
4- اندازهگیری فعالیت AAT سرم به روش ظرفیت مهاری تریپسین
5- تعیین فنوتیپهای آلفا-1-آنتیتریپسین به روش ایزوالکتروفوکوسینگ
سنجش فعاليت آلفا-1- آنتيتريپسين سرمي به روش ظرفيت مهاري تريپسين:
پروتئینهای آنتیتریپسینی سرم، هیدرولیزN بنزوئیل-DL- آرژینین-P-نیتروآنیلید (BAPNA) توسط تریپسین در بافر تریس را مهار میکنند و فعالیت AAT بر اساس میزان تریپسین مهارشده تعیین میگردد. برای این کار، سرم را با مقادیر مشخص تریپسین مخلوط کرده و در مرحله بعد به آن سوبسترای تریپسین اضافه ميشود و بر اساس شدت رنگ ایجاد شده، فعالیت AAT نمونه سرم تعیین ميشود.
سرولوپلاسمین (Ceruloplasmin)
سرولوپلاسمین، یک پروتئین (از نوع آلفا-2- گلبولین و یک گلیکوپروتئین) حاوی مس است که در غلظتهای بالاتر به عنوان یک آنزیم نیز عمل میکند. سرولوپلاسمین، یک فرواکسیداز بوده و با تبدیل آهن به فرم فریک، آن را جهت اتصال به ترانسفرین آماده میسازد. مس در واکنشهای متعددی شرکت میکند.
| Key copper-containing enzymes and their functions | |
| Enzymes | Function |
| Amine oxidases | Group of enzymes oxidizing primary amines (e.g., tyramine, histidine and polylamines) |
| Ceruloplasmin (ferroxidase I) | Multi-copper oxidase in plasma, essential for iron transport |
| Cytochrome c oxidase | Terminal oxidase enzyme in mitochondrial respiratory chain, involved in electron transport |
| Dopamine β-hydroxylase | Involved in catecholamine metabolism, catalyzes conversion of dopamine to norepinephrine |
| Hephaestin | Multi-copper ferroxidase, involved in iron transport across intestinal mucosa into portal circulation |
| Lysyl oxidase | Cross-linking of collagen and elastin |
| Peptidylglycine alpha-amidating mono-oxygenase (PAM) | Multifunction enzyme involved in maturation and modification of key neuropeptides (e.g., neurotransmitters, neuroendocrine peptides) |
| Superoxide dismutase (Cu, Zn) | Intracellular and extracellular enzyme involved in defense against reactive oxygen species (e.g., destruction of superoxide radicals) |
| Tyrosinase | Enzyme catalyzing melanin and other pigment production |
مس Copper (Cu2+)
مس، یک عنصر نادر (میکرومینرال) ضروری بوده که برای سنتز هموگلوبین و اکسیداسیون- احیاء موردنیاز میباشد. به صورت طبیعی، ادرار حاوی مقادیر بسیار کمی از مس آزاد بوده؛ در حالی که مس موجود در پلاسما اغلب به صورت متصل به سرولوپلاسمین میباشد. بررسی مس ادرار به منظور تشخیص بیماری ویلسون، مورد استفاده قرار میگیرد. در بیماری ویلسون، کاهش سطح سرولوپلاسمین که توسط کبد تولید میشود، موجب کاهش سطح مس موجود در پلاسما شده؛ در حالی که مس ادرار افزایش مییابد. درمان بیماری ویلسون از طریق تجویز پنیسیلامین صورت میگیرد که موجب افزایش میزان دفع کلیوی مس میگردد.
برای انجام این آزمایش، بیمار باید حداقل به مدت 1 هفته قبل از مصرف مکملهای ویتامینی، مواد معدنی و یا گیاهی خودداری نماید. برای انجام این آزمایش، جمعآوری ادرار 24 ساعته در ظرفی که حاوی یک نگهدارنده مناسب میباشد، لازم است. در طی مدت زمان جمعآوری ادرار 24 ساعته، ظرف حاوی ادرار باید در یخچال و یا فریزر نگهداری شود. مقدار طبیعی آن 60- 0 میکروگرم در ادرار 24 ساعته میباشد. مقدار طبیعی آن در افراد مسن، افزایش مییابد.
بیماری ویلسون (Wilson’s disease)
بیماری ویلسون (با شیوع 1 در 30 هزار نفر) که یک بیماری اتوزومال مغلوب میباشد، با کاهش سرولوپلاسمین سرم همراه میباشد. در این بیماری، یکی از متعدد جهشهای اتفاق افتاده، در ژن کدکننده ATP7B روی کروموزوم 13 میباشد که عضو جدیدی از خانواده ATPase نوع P انتقالدهنده کاتیون میباشد. این پروتئین در کبد سنتز شده و باعث ترشح مس به درون پلاسما میگردد. بیش از 200 جهش از ژن بیماری ویلسون که منجر به اختلال عملکرد ATP7B و تجمع داخل سلولی مس میشود، شناسایی شده است.
مس اضافی در درون لیزوزومهای هپاتوسیتها رسوب کرده و واکنشهای رادیکال آزاد شامل پراکسیداسیون لیپیدی و ناپایداری غشایی را القاء میکنند که میتواند به هپاتیت فعال مزمن، سیروز و یا نارسایی برقآسای کبدی منجر شود. مس در سیستم عصبی مرکزی به ویژه هسته لنتیکولار بازال گانگلیا رسوب کرده و ایجاد اختلال روانی مینماید. همچنین مس میتواند در اطراف عنبیه رسوب کرده و حلقههای کایزر- فلشر (Keyser-Fleischer) را تشکیل دهد.
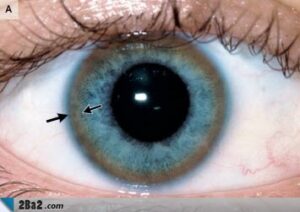
وجود حلقه کایزرفلشر (Keyser-Fleischer) (یک حلقه قهوهای در اطراف عنبیه) در بیماری ویلسون، به ویژه در هنگامی که علائم عصبی وجود دارند، شایع میباشد
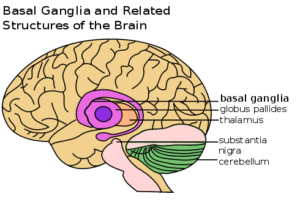
محل بازال گانگلیا (basal ganglia) در مغز که در بیماری ویلسون تحتتأثیر قرار میگیرد
تشخیص بیماری ویلسون بر پایه علایم بالینی و یافتههای آزمایشگاهی استوار است که شامل کاهش سرولوپلاسمین و مس سرم، افزایش ترشح ادراری مس و افزایش محتوای مس کبد میباشد. فاکتورهای افزایشدهنده سنتز سرولوپلاسمین (مانند سیتوکاینها، حاملگی و استروژنها) ممکن است باعث ایجاد مقادیر طبیعی سرولوپلاسمین در 15 درصد از کل بیماران و 35 درصد از بیماران با تظاهرات کبدی بیماری ویلسون، به ویژه هپاتیت ویلسونی گردند. حضور 250 میکروگرم از مس در هر گرم از بافت خشک کبد، تأییدکننده بیماری ویلسون میباشد. تست ژنتیکی، معتبرترین روش اثبات این بیماری بوده، اما به دلیل این که بیش از 200 جهش مؤثر در ایجاد این بیماری نقش دارند، مشکل میباشد.
درمان: بیماران باید از مصرف غذاهای سرشار از مس مانند قارچ، شکلات، میوههای خشک، جگر و … خودداری نمایند. داروی پنیسیلامین از طریق اتصال به مس (شلاته کردن) و دفع آن در ادرار عمل مینماید. افرادی که نمیتوانند پنیسیلامین را تحمل کنند، میتوانند از داروی با اثر مشابه دیگر، Trientine hydrochloride استفاده کنند. پس از برگشت نتایج به حد طبیعی، ممکن است که به جای شلاتهکنندهها، از روی به فرم استات روی (Zinc acetate) استفاده شود که موجب تحریک متالوتیونین شود که یک پروتئین در سلولهای روده بوده که از طریق اتصال به مس، مانع جذب و انتقال آن به کبد میگردد.
بیماری منکه (Menkes disease)
بیماری منکه که به اسامی Steely hair disease و Kinky hair disease نیز نامیده میشود، یک بیماری مغلوب وابسته به کروموزوم X میباشد.
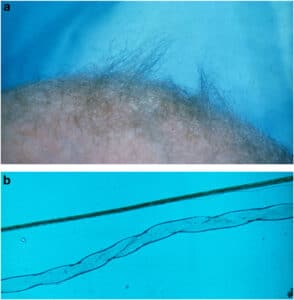
موی غیرطبیعی در یک بیمار مبتلا به بیماری منکه. تصویر a: ظاهر زبر موهای بدون رنگدانه در سر. تصویر b: مقایسه ظاهر میکروسکوپی موی طبیعی (بالا) با موی پیچخورده (پایین) در بیماری منکه
در این بیماری، جهش در ژن کدکننده ATP7A روی کروموزوم X عامل بیماری میباشد. در نتیجه این موتاسیون، حمل و نقل مس در رژیم غذایی از سلولهای روده، دچار اختلال شده که منجر به تجمع مس در بافتهایی از قبیل روده کوچک و کلیهها و غلظت پایین مس در سرم میشود.
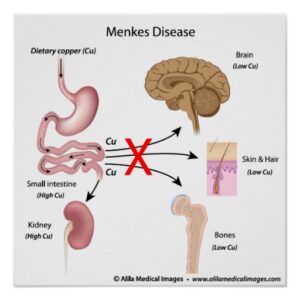
تشخیص: نسبت همووانیلیک اسید به وانیلیل ماندلیک اسید ادرار (Urine homovanillic acid/vanillylmandelic acid ratio) برای بررسیهای اولیه به کار میرود.
درمان: تزریق زیرجلدی و یا درون رگی مکملهای مس به فرم نمکهای استات، ممکن است در این بیماران مفید باشد.
فاکتورهای انعقادی
پروتئینهای انعقادی بجز فونویلبراند که توسط سلولهای اندوتلیال و مگاکاریوسیتها ساخته میشود، در کبد ساخته میشوند. مهارکنندههای انعقادی از قبیل آنتیترومبین III، آلفا-2- ماکروگلبولین، آلفا-1- آنتیتریپسین، مهارکننده C1 استراز و پروتئین C نیز در کبد ساخته میشوند. مقادیر پایین آنتیترومبین III در بیماران مبتلا به سیروز و هپاتیت، ممکن است در نتیجه کاهش سنتز، افزایش مصرف و یا تغییر در میزان عبور مویرگی ایجاد گردد. شایعترین اختلال انعقادی مشاهده شده در نارسایی کبدی، یعنی سیروز و نارسایی برقآسای کبدی، انعقاد داخل عروقی منتشر (DIC) میباشد.
این بیماری با افزایش مصرف فاکتورهای انعقادی و پلاکتها مشخص شده و باعث ترومبوسیتوپنی و افزایش زمان PT و PTT میگردد. تشخیص DIC میتواند به وسیله تعیین مقادیر افزایش یافته D- دایمر خون، قطعی گردد. در برخی موارد نارسایی کبدی، تعداد پلاکتها به دلیل محبوس شدن در یک طحال بزرگ به علت هپاتواسپلنومگالی، کاهش مییابد. این حالت به همراه مقادیر افزایش یافته PT و PTT ناشی از کاهش سنتز فاکتورهای انعقادی میتواند تظاهرات DIC را تقلید نماید. در این گونه موارد، D-دایمر افزایش نیافته و تشخیص DIC را رد مینماید.
امتیاز MELD (Model for end-stage liver disease)
PT یکی از اجزای امتیازبندی MELD بوده که به منظور ارزیابی ضرورت پیوند کبد در بیماری کبدی به کار میرود. این امتیاز، یک نمره محاسبه شده بر اساس مقادیر بیلیروبین، کراتینین و INR (International Normalized Ratio) میباشد. این نمره ظاهراً به طور دقیقی مرگ 3 ماهه را برای بیماران سیروزی منتظر پیوند کبدی، پیشبینی میکند.
MELD بر اساس فرمول زیر محاسبه میشود:
MELD=3.78×ln[bilirubin(mg/dL)]+11.2×ln[INR]+9.57×ln[creatinine(mg/dL)]+6.43×aetiology(0:cholestatic or alcoholic, 1- otherwise)
| امتیاز MELD | احتمال مرگ در 3 ماه آینده |
| 40 ≤ | 71/3% |
| 39- 30 | 52/6% |
| 29- 20 | 19/6% |
| 19- 10 | 6% |
| 9 > | 1/9% |
دس- گاما- کربوکسی پروترومبین (Des-gamma-carboxy prothrombin; DCP)
فاکتورهای انعقادی وابسته به ویتامین K (II ,VII ,IX ,X) در کبد سنتز شده و برای پردازش پس از ترجمه خود، نیاز به ویتامین K (برای گاماکربوکسیلاسیون تعدادی از واحدهای اسید گلوتامیک انتهایی به گاماکربوکسی گلوتامیک اسید) دارند که قبل از ترشح به خون صورت گرفته و برای عملکرد فعال این فاکتورها در آبشار انعقادی ضروری میباشد. پیشساز پروترومبین بدون پردازش (DCP)، در یک سری از بیماران مبتلا به کارسینومای هپاتوسلولار افزایش مییابد که پیشبینی کننده کاهش میزان بقای بیمار میباشند. این آزمون میتواند به عنوان یک تومورمارکر مفید در تشخیص زودرس بیماری کبد مورد استفاده قرار گیرد .
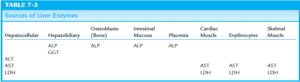
آنزیمهای کبدی
| محل وجود آنزیم | مثال | علت افزایش در خون |
| سیتوپلاسم سلولها | LDH، AST و ALT | رها شدن به خون در اثر آسیب به غشای سلولهای هپاتوسیتی |
| میتوکندری | AST | رها شدن در اثر مصرف اتانول |
| مجاری کبدی و صفراوی | ALP، GGT و 5ʹ-N | رها شدن در اثر انسداد مجاری صفراوی و تجمع نمکهای صفراوی. افزایش سنتز GGT و به میزان کمتری ALP، به واسطه مصرف داروهایی از قبیل اتانول، فنیتوئین و کاربامازپین از طریق القاء سنتز آنزیمهای میکروزومی |
آمینوترانسفرازها (ترانسآمینازها) (SGOT & SGPT)
ترانسآمینازها (آمینوترانسفرازها) شامل AST (آسپارتات آمینو ترانسفراز) و ALT (آلانین آمینوترانسفراز) میباشند. اسم قدیمی AST، SGOT (گلوتامات اگزالواستات ترانسآمیناز سرمی) و اسم قدیمی ALT، SGPT (گلوتامات پیروات ترانسآمیناز سرمی) میباشد.
ترانسآمینازها در انتقال برگشتپذیر گروه آمین یک اسید آمینه به آلفاکتوگلوتارات و تشکیل گلوتامات و آلفاکتواسید مربوطه به همان اسید آمینه، نقش دارند.
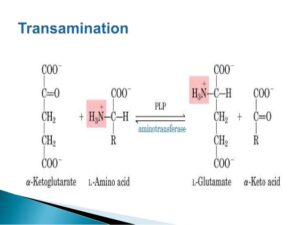
AST در انتقال برگشتپذیر گروه آمین اسیدآمینه آسپارتات به آلفاکتوگلوتارات و تشکیل گلوتامات و اگزالواستات، نقش دارد.
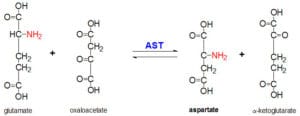
ALT در انتقال برگشت پذیر گروه آمین اسیدآمینه آلانین به آلفاکتوگلوتارات و تشکیل گلوتامات و پیروات، نقش دارد.
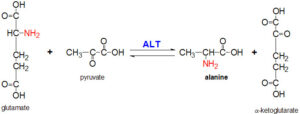
آمینوترانسفرازها به ویتامین B6 به فرم PLP به عنوان کوفاکتور نیاز دارند. در اکثر آزمایشهای سرمی مربوط به ALT و AST، چنین در نظر گرفته میشود که سرم بیمار، PLP موردنیاز برای واکنش را تأمین میکند. نقص B6 در الکلیها، شایع میباشد.
| آنزیم | نیمه عمر | محل وجود آنزیم | حضور در بافتها | میزان تام سیتوپلاسمی نسبت به پلاسما |
| AST | 17 ساعت
(فرم میتوکندریایی 87 ساعت) |
سیتوپلاسم و میتوکندری | قلب، ماهیچه و کبد | 7000 برابر پلاسما |
| ALT | 47 ساعت | سیتوپلاسم | کبد و مقداری در کلیهها | 3000 برابر پلاسما |
به نسبت AST به ALT، نسبت DeRitis گفته میشود. در حالت طبیعی این دو آنزیم به نسبت 1 به 1 وجود دارند و بنابراین در حالت طبیعی، نسبت AST/ALT برابر 1 خواهد بود.
در آسیب حاد هپاتوسلولار (نظیر هپاتیت)، AST به خاطر نسبت بیشترش (7000 برابر پلاسما)، اساساً بیشتر از ALT خواهد بود (AST/ALT >1). در عرض 48- 24 ساعت، به ویژه اگر آسیب ناگهانی اتفاق بیفتد، ALT به دلیل داشتن نیمه عمر بالاتر، بیشتر از AST خواهد شد (AST/ALT <1) (البته به استثنای هپاتیت الکلی).
الکل با القاء آسیب میتوکندریایی، باعث افزایش نامتناسب AST به ALT شده و نسبتی از AST به ALT را به میزان 4- 3 به 1 ایجاد مینماید.
در آسیب مزمن هپاتوسیتها (عمدتاً سیروز)، معمولاً نسبت ALT به AST افزایش مییابد. با این حال، با پیشرفت فیبروز، فعالیت ALT معمولاً کاهش یافته و نسبت AST اغلب بالاتر از ALT خواهد بود. در مراحل نهایی سیروز، مقادیر هر دو آنزیم عموماً افزایش نیافته و ممکن است در نتیجه تخریب وسیع بافتی، دچار کاهش شوند.
AST برای کنترل درمان با داروهای هپاتوتوکسیک به کار میرود و یک AST بیش از 3 برابر حد طبیعی، علامت توقف درمان میباشد. افزایش مزمن فعالیتهای آمینوترانسفرازی در بیماران بدون علامت، ممکن است علتهای متعددی نظیر استفاده از الکل یا دارو، هپاتیت مزمن ویروسی یا بیماری کبد چرب غیرالکلی داشته باشد.
اندازهگیری: با افزودن آلانین به ALT و یا آسپارتات به AST، گلوتامات تولید شده که تحتتأثیر گلوتامات دهیدروژناز، تولید آلفاکتوگلوتارات مینماید. در این واکنش، NAD به NADH تبدیل شده که به صورت یک افزایش جذب در 340 نانومتر قابل اندازهگیری است.
لاکتات دهیدروژناز Lactic Dehydrogenase; Lactate Dehydrogenase (LDH; LD)
لاکتات دهیدروژناز (LDH) آنزیمی است که به طور عمده در قلب، کبد، عضلات اسکلتی و گلبولهای قرمز یافت میشود. همچنین این آنزیم به مقادیر کمتر در مغز، کلیه، ریه، پانکراس و طحال نیز یافت میشود. LDH شامل تترامرهایی از دو شکل H (دارای تمایل بالا برای لاکتات) و M (دارای تمایل بالا برای پیروات) میباشد و اکسیداسیون قابل برگشت لاکتات به پیروات را کاتالیز میکند.
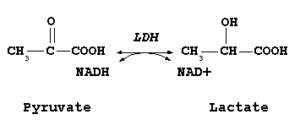
LDH میتواند به صورت توتال و یا به صورت هر کدام از ایزوآنزیمهای 5گانهی آن اندازهگیری شود. اندازهگیری ایزوآنزیمهای آن می تواند به افتراق و تشخیص بافتی که مسئول افزایش LDH میباشد، کمک نماید.
| Type | Composition | Location |
| LDH1 | HHHH | Heart and Erythrocyte |
| LDH2 | HHHM | Heart and Erythrocyte |
| LDH3 | HHMM | Brain and Kidney |
| LDH4 | HMMM | Skeletal Muscle and Liver |
| LDH5 | MMMM | Skeletal Muscle and Liver |
LDH به همراه آسپارتات آمینوترانسفراز (AST) و کراتینکیناز (CK) معمولاً در موارد شک به سکته قلبی (MI) ارزیابی میشوند؛ اگرچه اکنون با وجود آزمایش تروپونین، استفاده از LDH برای تشخیص سکته قلبی کمتر شده است. LDH تقریباً 12 ساعت بعد از آسیب بافتی در خون پدیدار شده و در 48-24 ساعت پس از ایجاد آسیب، به حداکثر مقدار خود میرسد. مقدار ماکزیمم LDH، متعاقب سکته قلبی ممکن است به 800-300 واحد در لیتر برسد. سطح LDH تقریباً به مدت 10 روز بالا باقی خواهد ماند. در حالت عادی و در افراد طبیعی، سطح LD2 بیشترین درصد از LDH توتال را به خود اختصاص میدهد؛ در حالی که در طی سکته قلبی، میزان LD1 نسبت به LD2 افزایش مییابد (Flipped ratio).
مقادیر سرمی LDH در هپاتیتها افزایش مییابد که اغلب گذرا بوده و با گذشت زمان به حالت طبیعی برمیگردد. افزایش LDH تام تا مقادیر IU/L 500 یا بیشتر (محدوده مرجع IU/L 150)، توأم با یک افزایش قابل ملاحظه در ALP تا مقادیر بیش از IU/L250، در غیاب سایر ناهنجاریهای آنزیمی به ویژه AST و ALT، حائز اهمیت است. این افزایش انتخابی، اغلب با ضایعات پیشرونده کبدی نظیر کارسینومای متاستاتیک، کارسینومای هپاتوسلولار اولیه و یا به طور نادری، ضایعههای خوشخیمی نظیر همانژیوما همراه میباشد. LD5 میتواند از هپاتوسیتها، تومورها و یا هر دو ناشی شود.
LDH در 4 درجه سانتيگراد به علت حساسیت LD5 به سرما، پایدار نمیباشد. بستن گارو میتواند مقدار آنزیم را شدیداً افزایش دهد. تغییرات روزانه در مردان وجود ندارد، اما در زنان، فعالیت آنزیم در عصرها تا 40% بیشتر از صبح است. بعد از ورزش مقدار LDH ممکن است تا 50% افزایش یابد.
مقدار طبیعی لاکتات دهیدروژناز توتال 210-110 واحد در لیتر میباشد. مقدار طبیعی ایزوآنزیمهای LDH1 17-27%، LDH228-38%، LDH3 17-28%، LDH4 5-15% و LDH5 5-15% میباشد. مقادیر LDH در نوزادان و شیرخواران بیشترین مقدار را دارد.
آلکالن فسفاتاز Alkaline Phosphatase (ALP)
آلکالن فسفاتاز (ALP)، آنزیمی است که در کبد، استخوانها، جفت، رودهها و کلیهها یافت میشود. بیشترین غلظت این آنزیم در سلولهای پوشاننده مجاری صفراوی و در استئوبلاستها (سلولهای درگیر در تشکیل استخوانهای جدید) یافت میشود. ALP به طور طبیعی از کبد به مجاری صفراوی ترشح میشود. افزایش سطح ALP به طور عمده در طی دوره رشد استخوانها (مثلاً در کودکان)، در انواعی از بیماریهای کبدی و در انسداد مجاری صفراوی مشاهده میشود.
ALP همچنین به عنوان یک تومور مارکر در تشخیص Osteogenic sarcoma (Osteosarcoma) و همچنین سرطانهای کبد و پروستات که به استخوان متاستاز داده باشند، به کار میرود. استئوژنیک سارکوما یک نئوپلاسم اولیه بدخیم استخوانها میباشد که متشکل از یک استرومای بافت همبندی بدخیم همراه با استئوئید، استخوان و یا غضروف بدخیم است. زیرگروههای این نئوپلاسم شامل استئوبلاستیک، کندروبلاستیک و یا فیبروبلاستیک میباشند.
هر کدام از بافتهای حاوی ALP، دارای ایزوآنزیمهای مجزایی بوده که توسط الکتروفورز از یکدیگر تفکیک میشوند.
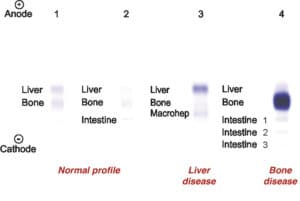
ALP تام سرم، عمدتاً به فرم غیرمتصل و به میزان کمتری، به صورت کمپلکس با لیپوپروتئینها و به ندرت، ایمونوگلبولینها وجود دارد. ALP مربوط به روده کوچک و جفت، از لحاظ آنتیژنی، متفاوت از فرمهای کبدی، کلیوی و استخوانی هستند.
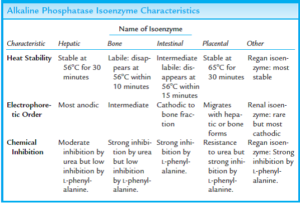
ALP کبدی و استخوانی، ALP سرم بیماران عادی را تشکیل میدهد. مقدار طبیعی آن در خانمها، 100-30 واحد در لیتر و در آقایان، 115-45 واحد در لیتر میباشد. مقدار طبیعی آن در بچهها، 3-1 برابر افراد بالغ میباشد. مقدار طبیعی آن در سنین بلوغ نیز 6-5 برابر افراد بالغ میباشد. مقدار طبیعی آن در افراد مسن، مختصری بالاتر از افراد بالغ است.
گاماگلوتامیل ترانسفراز (GGT)
آنزیم گاماگلوتامیل ترانسفراز (GGT) در انتقال اسیدهای آمینه و پپتیدها از غشاء سلولی و احتمالاً در متابولیسم گلوتاتیون شرکت دارد. بیشترین غلظت این آنزیم در کبد و مجاری صفراوی وجود دارد. در بیماریهای کبدی، همانند لوسین آمینوپپتیداز و ´5ـ نوکلئوتیداز، افزایش سطح گاماگلوتامیل ترانسفراز معمولاً موازی با افزایش سطح آلکالن فسفاتاز رخ میدهد؛ اگرچه، معمولاً حساسیت گاماگلوتامیل ترانسفراز، بیشتر است. برعکس، در موارد بیماریهای استخوانی، همانند لوسین آمینوپپتیداز و ´5ـ نوکلئوتیداز، سطح گاماگلوتامیل ترانسفراز افزایش نمییابد و در این موارد، تنها سطح آلکالن فسفاتاز افزایش مییابد.
به عبارتی سادهتر، افزایش همزمان آلکالن فسفاتاز و گاماگلوتامیل ترانسفراز، نشاندهنده بیماریهای کبدیـ صفراوی بوده، در حالی که افزایش سطح آلکالن فسفاتاز در حضور مقادیر طبیعی گاماگلوتامیل ترانسفراز، نشاندهنده بیماریهای استخوانی میباشد. لازم به ذکر است که تركيب آلكالن فسفاتاز بالا و گاماگلوتامیل ترانسفراز طبيعي، به طور كامل ردكننده بيماري كبدي نيست، زیرا در مواردی مانند اواخر حاملگي ممکن است سطح گاماگلوتامیل ترانسفراز كاهش يابد.
استفاده دیگر گاماگلوتامیل ترانسفراز در تشخیص مصرف مزمن الکل میباشد، بنابراین، این آزمایش ابزار مفیدی در غربالگری و بررسی بیماران الکلی میباشد. افزایش گاماگلوتامیل ترانسفراز تقریباً در 70% بیمارانی که به صورت مزمن، الکل مصرف میکنند، مشاهده میشود.
گلوتاتیون، توانایی عبور از غشای سلولی و ورود به درون سلول را نداشته، بنابراین گلوتاتیون توسط GGT به اسیدهای آمینه سازنده خود تجزیه شده و پس از عبور از غشای سلولی، در درون سلول، مجدداً به یکدیگر پیوسته و تشکیل گلوتاتیون را میدهند.
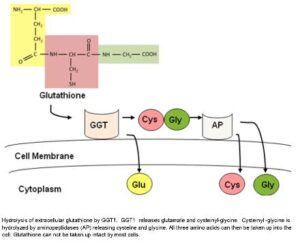
گلوتاتیون برای حفاظت از سلولها در برابر مواد اکسیدان که حتی در طی متابولیسم طبیعی نیز تولید میشوند، موردنیاز میباشد. متعاقب ایجاد استرس اکسیداتیو و افزایش مواد اکسیدان در درون سلولها، نیاز به افزایش گلوتاتیون مشاهده میشود. همچنین تولید رادیکالهای آزاد که میتواند با شدت بیشتر در افراد با کبد چرب و چاقی شکمی رخ دهد، سبب خالی شدن محتوای گلوتاتیون داخل سلولی و در نتیجه، القاء فعالیت GGT و افزایش آن در داخل گردش خون میشود. اخیراً از GGT به عنوان یک مارکر در پیشگویی بیماریهای قلبی ـ عروقی، دیابت نوع II و… استفاده میشود.
اندازهگیری: اکثر آزمایشهای مربوط به GGT از گاماگلوتامیل پارانیتروآنیلید به عنوان سوبسترا استفاده میکنند. در این واکنش، پارانیتروآنیلین آزاد شده، رنگزا بوده که از طریق اسپکتروفتومتری قابل اندازهگیری میباشد.
ʹ5- نوکلئوتیداز 5ʹ- Nucleotidase (5ʹ- N)
آزمایش ʹ5- نوکلئوتیداز (5ʹ-N) در ارتباط با آلکالن فسفاتاز (ALP) به منظور افتراق بین بیماریهای مجاری صفراوی و بیماریهای استخوانی مورد استفاده قرار میگیرد. آنزیم ʹ5- نوکلئوتیداز در غشای سلولهای کبدی و سلولهای مجاری صفراوی یافت میشود. به دلیل محدود بودن مکان تولید این آنزیم، اختصاصیت این آنزیم نسبتاً زیاد میباشد. در مواقعی که 5ʹ-N و ALP هر دو افزایش مییابند، احتمال متاستاز به کبد نیز میتواند مطرح باشد.
مقدار طبیعی ʹ5- نوکلئوتیداز 11-1 واحد در لیتر میباشد.
پرهآلبومین Prealbumin (PAB)
پرهآلبومین یک پروتئین پلاسمایی است که در کبد ساخته شده و از آن به عنوان بهترین مارکر برای سوءتغذیه یاد میشود. نیمهعمر پرهآلبومین در حدود 2 روز بوده و از این رو، تغییرات سطح آن به سرعت رخ داده و منعکس کننده وضعیت تغذیهای اخیر شخص میباشد. از این آزمایش اغلب در تشخیص سوءتغذیه پروتئین- انرژی استفاده میشود. ارزیابی سطح پرهآلبومین در بیماران دارای بیماریهای مزمن، بیماران بستری شدهی دارای ریسک بالا و قبل از بسیاری از عملهای جراحی، امری ضروری و مهم میباشد. تشخیص و اصلاح کمبود تغذیهای میتواند به جلوگیری از بروز عوارض آن و بهبود وضعیت بیمار کمک نماید.
این آزمایش همچنین در ارزیابی بهبود وضعیت بیمارانی که تغذیهی حمایتی از قبیل تغذیهی وریدی دریافت میکنند نیز به کار میرود. بیماران دارای سطوح پایین پرهآلبومین نیازمند مشاوره تغذیهای و اصلاح کمبودهای تغذیهای میباشند. بیماران دارای سطوح پایین پرهآلبومین ممکن است دارای کاهش در سطح سایر پروتئینها نیز باشند.
لوسین آمینوپپتیداز Leucine Aminopeptidase (LAP)
لوسین آمینوپپتیداز (LAP) آنزیمی است که به طور طبیعی در سلولهای کبدی (هپاتوسیتها) یافت شده و در خون، صفرا و ادرار وجود دارد. این آنزیم، پس از آسیب سلولهای کبدی متعاقب مصرف داروهای دارای اثر سمی بر کبد و یا عفونتها از قبیل هپاتیت، به درون خون آزاد میشود. لوسین آمینوپپتیداز همچنین ممکن است توسط تومورهای موجود در کبد به درون خون رها شده و از این رو ممکن است همانند یک تومور مارکر در نظر گرفته شود. این آزمایش در تشخیص مواردی که با افزایش سطح آلکالن فسفاتاز (ALP) همراه هستند، مفید میباشد. تغییرات سطح لوسین آمینوپپتیداز (LAP)، همسو با تغییرات سطح آلکالن فسفاتاز (ALP) بوده، بجز در مواردی که با بیماریهای استخوانی و یا سوء جذب مواجه هستیم، که در این مورد، سطح لوسین آمینوپپتیداز (LAP) طبیعی میباشد.
لوسین آمینوپپتیداز (LAP) اغلب حساسیت سایر آزمایشهای کبدی از قبیل ALT، AST، ALP، LDH و GGT را ندارد. از مزایای لوسین آمینوپپتیداز (LAP) این است که برخلاف سایر آنزیمهای کبدی، میتوان آن را در ادرار اندازهگیری نمود. مقدار طبیعی لوسین آمینوپپتیداز در خانمها 185-75 واحد در میلیلیتر و در آقایان 200-80 واحد در میلیلیتر میباشد.
محل وجود آنزیمهای کبدی به طور خلاصه
آلفافتوپروتئین (AFP)
آلفافتوپروتئین (AFP) توسط هپاتوسیتها و سلولهای کیسه زرده جنینی تولید میشود و در 3 ماهه دوم حاملگی، به حداکثر مقدار خود میرسد. ممکن است AFP، یک سرکوبکننده سیستم ایمنی باشد که از تخریب بافتهای جنینی توسط آنتیبادیهای در گردش خون مادری جلوگیری میکند. AFP در نقایص لوله عصبی جنینی (NTDs) تا سطح غیرطبیعی شروع به افزایش میکند. مقادیر طبیعی AFP با سن حاملگی تغییر میکند و بنابراین باید در هنگام آزمایش و تفسیر آن، این نکته مدنظر قرار گیرد. اندکی پس از تولد، مقدار AFP کاهش یافته و در حدود 1 سالگی به حد طبیعی بزرگسالی میرسد. در آسیب حاد کبدی، یک افزایش در AFP (معمولاً ng/dl100- 200) به دلیل تولید دوباره آن توسط هپاتوسیتها اتفاق میافتد.
AFP مارکر مهمی برای کارسینومای هپاتوسلولار (HCC) میباشد. مقادیر افزایش یافته AFP در بیش از 90% بیماران مبتلا به HCC، مشاهده شده است. افزایش مقادیر AFP پس از بیماری حاد کبدی و نیز فیبروز، گاهی اوقات این مارکر را غیراختصاصی میکند، با این حال، در مقادیر بیشتر از ng/dl 400، احتمال بالایی از HCC وجود دارد.
از AFP در تستهای غربالگری سلامت جنین مانند تریپل مارکر، کواد مارکر و پنتا مارکر نیز استفاده میشود.
تریپل مارکر: AFP + hCG + uE3
کواد مارکر: AFP + hCG + uE3 + (DIA) Inhibin A
پنتا مارکر: AFP + hCG + uE3 + (DIA) Inhibin A + (ITA) Invasive Trophoblast Antigen
منابع:
1-Tietz Textbook of Clinical Chemistry and Molecular Diadnosis. 2006; 4th Edition.
2- Henrys Clinical Diagnosis and Management by Laboratory Methods. 2007; 21st Edition.
3 – Wendy Aineson and Jean Brickell. Clinical Chemistry; A Laboratory Perspective. 2007; 1st Edition.
4- Arneson W, Brickell J. Clinical chemistry; a laboratory perspective. 2007.
5- Pagana KD and Pagana TJ. Diagnostic and laboratory test refrence. 2005; 7th Edition.
6- Van Leeuwen AM, Kranpitz TR and Smith L. Laboratory and diagnostic tests with nursing implications. 2006 ; 2nd Edition.
7- Wilson DD. Manual of laboratory and diagnostic tests. 2008.
8- مراد رستمی و معصومه جرفی. شاخص PGA (PGA Index). مجله تشخیص آزمایشگاهی. مهرماه 1393، شماره 105، صفحه 9
گاما گلوتامیل ترانسفراز (Gamma Glutamyl transferase; GGT; c)
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام