کم خونی آپلاستیک
دکتر حبیباله گل افشان1، پریسا تندل2
1. هیئت علمی دانشگاه علوم پزشکی شیراز
2. کارشناسی ارشد خون شناسی آزمایشگاهی و بانک خون دانشکده پیراپزشکی دانشگاه علوم پزشکی شیراز
پانسایتوپنی به همراه مغز استخوان هایپوسلولار در غیاب غدد لنفاوی بزرگ یا طحال و کبد بزرگ مطرحکننده آنمی آپلاستیک (Aplastic Anemia) یا به اختصار AA است. البته متعاقب شیمیدرمانی و رادیوتراپی و تجویز سرکوبگرهای مغز استخوان، تصویر موقتی شبیه به آنمی آپلاستیک (AA) ایجاد میشود. در این موارد، فعال شدن بافت خونساز با منوسیتوز و میل به چپ در سری گرانولوسیتی همراه بوده و حتی ممکن است شاهد سلولهای بلاست در خون محیطی باشیم که پس از مدتی ناپدید میگردد. گفتنی است که در نوزادان و مواردی از قبیل الکلیسم که ذخیره ناکافی از گرانولوسیتها در مغز استخوان وجود دارد، یک استرس یا عفونت میتواند بهطور موقت میل به چپ شدید شبیه لوسمی حاد ایجاد کند.
کم خونی آپلاستیک (AA) نارسایی مغز استخوان است که با نارساییهای کلونال دیگر مغز استخوان مانند سندرم مایلودیسپلاستیک (MDS)، هموگلوبیناوری حملهای شبانه (PNH) و لوسمی لنفوسیتهای بزرگ دانهدار (T-LGL) همپوشی دارد و ممکن است به AML/MDS تبدیل شود.
برای شناسایی کم خونی آپلاستیک حداقل دو معیار از سه معیار Hb<10 و Platelet<50000 و ANC<1500 بایستی صادق باشد و بر اساس معیارهای فوق AA به سه گروه خیلی شدید، شدید و غیرشدید طبقهبندی میگردد.
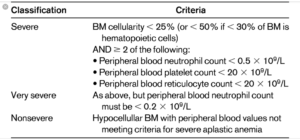
سلولاریتهی مغز استخوان کمتر از 25 درصد با 500>AGC و پلاکت کمتر از 20000 در میلیمتر مکعب و شمار تصحیحشدهی رتیکولوسیت کمتر از یک درصد، کمخونی را در گروه شدید قرار میدهد. چنانچه باوجود شرایط یادشده، تعداد خالص نوتروفیل کمتر از 200 در میلیمتر مکعب باشد، در گروه کمخونی بسیار شدید طبقهبندی میشود
شیوع AA در آسیا 2 تا 3 برابر نسبت به غرب است. ابتلای دو فازه در سنین 10 تا 25 سالگی و بالای 60 سال مشاهده میشود و تفاوتی بین ابتلای زن و مرد وجود ندارد.

پانسایتوپنی شدید در یک بیمار مبتلا به کمخونی اپلاستیک با شمارش رتیکولوسیت کمتر از نیم در صد. فقدان گلبولهای پلیکروماژی و نبود پلاکتهای بزرگ و توپر و نبود گلبولهای قرمز هستهدار در خون محیطی با تصویر پانسیتوپنی، احتمال کم خونی اپلاستیک را مطرح میکنند
حدود 5 تا 10 درصد از موارد کم خونی آپلاستیک پس از هپاتیت رخ میدهد. کمخونی پس از 6 هفته از ژاندیس و التهاب کبد شروع میشود و در این برهه ممکن است سرولوژی ویروس منفی شود. شدت کمخونی در جنس مذکر است و افزایش فعالیت لنفوسیتهای T سایتوتوکسیک (T-Cytotoxic) در اتیولوژی مطرح شده است و به سرکوبگرهای ایمنی (Immunosuppressive) یا به اختصار IST پاسخ میدهد. کم خونی اپلاستیک ممکن است در زمینهی بیماریهای اتوایمون، مانند لوپوس، کمخونی آپلاستیک در زمینه بیماریهای آتوایمیون بهویژه التهاب غلاف ائوزینوفیلیک Eosinophilic Fasciitis، لوپوس سیستمیک، روماتیسم مفصلی، سندرم شوگرن و بیماری سیلیاک و بیماریهای آتوایمون تیروئید ممکن بهندرت رخ دهد. در لوپوس گاهی پانسایتوپنی به همراه فیبروز مغز استخوان (Autoimmune Fibrosis) همراه است.
تیموما (Thymoma) گرچه در اکثر موارد با آپلازی خالص گلبولهای قرمز همراهی دارد، اما ممکن است با کمخونی آپلاستیک نیز ظاهر شود.
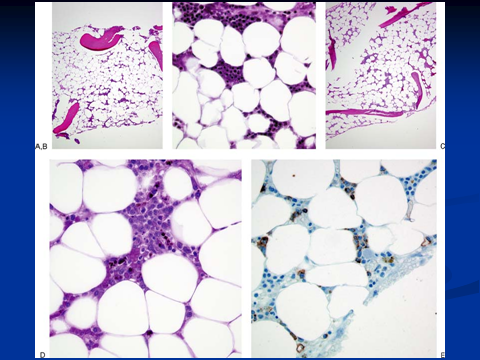
کمخونی اپلاستیک ممکن است بهطور ناگهانی یا بهتدریج، در طول هفتهها یا ماهها بروز کند. ویژگی مهم کم خونی اپلاستیک مغز استخوان هیپوسلولار با سلولاریته کمتر از 25درصد در بیوپسی مغز استخوان است. خون محیطی با نمای پانسیتوپنی و نبود تغییرات دیسپلاستیک و سلول غیرطبیعی تشخیص را تأئید میکند
کم خونی آپلاستیک بسیار مرگآور در افراد با کاهش ایمنی سلولار رخ میدهد که اخیراً تزریق خون غیر اشعه دادهشده (non irradiated) داشتهاند. بیماران با کاهش ایمنی سلولار، لنفوم هوچکین، بیماری که شیمیدرمانی با آنالوگهای پورین میشود و بیماری که از بستگان درجه اول خون دریافت میکند، همگی مستعد واکنش گرافت در رابطه با تزریق خون هستند؛ بدین مفهوم که لنفوسیتهای زنده T در کیسه خون پس از تزریق به بافتهای پوست، گوارش، کبد و مغز استخوان تهاجم داده و عوارضی از قبیل درماتیت، گاستروانتریت، هپاتیت و کمخونی آپلاستیک را بوجود میآورد که درمان نداشته و موجب مرگ بیمار میشود.
در همه موارد فوق، کیسههای خون یا پلاکت بایستی پس از دریافت 2500 راد اشعه گاما به بیمار تزریق شود. اشعه گاما و ایکس موجب مرگ و غیرفعال شدن لنفوسیتهای T گردیده و از سندرم گرافت جلوگیری میکنند. بیماران مبتلا به کمخونی آپلاستیک که داروی ATG (antithymocyte globulin) میگیرند نیز کاندیدای تزریق خون اشعهدیده هستند.
همراهی کمخونی آپلاستیک با بیماریهای آتوایمیون و لوسمی T-LGL با تکثیر کلونال لنفوسیتهای +CD8 از دیرباز شناخته شده است و در حدود 7% بیماران آپلاستیک جهش 3 STAT در سلولهای T مشاهده شده که با ایجاد بینظمی در پاسخ ایمنی موجب کمخونی آپلاستیک میگردد.
کمخونی آپلاستیک بهندرت در حاملگی مشاهده شده است. پیوند آلوژنیک موفقیتآمیز موجب عود کمخونی در حاملگیهای بعد نمیشود. لیست بزرگی از داروها در ایجاد آنمی آپلاستیک متهم هستند که با سازوکارهای ایمونولوژیک موجب تخریب مغز استخوان میگردند. کمخونی آپلاستیک با اختلال کمی و کیفی در حوضچه سلولهای مادر خونساز با طول عمر طولانی (Long term) و بالغتر که در چرخه تولید انواع سلولهای خون هستند همراه است. فقدان یا کاهش سلولهای مادر و مستعد آپوپتوز بودن آنها با آنالیز ترانسکریپتوم (Transcriptome) مشاهده شده است.
حدود 10 تا 15% بیماران مبتلا به AA دارای تلومر کوتاه در لکوسیتهای خون هستند. مطالعات، اختلال چشمگیری در ساختار ریزمحیطی و فاکتورهای رشد خونساز در کمخونی را نشان نداده است. اشعهی یونیزان، بهطور مستقیم برای سلولهای مادر خونساز و سلولهای اجدادی، کشنده است. سلولهای مادر خونساز و لنفوسیتها در برابر آسیب ناشی از اشعه بسیار حساساند؛ درحالیکه حساسیت گرانولوسیتها و مگاکاریوسیتها متوسط است. پلاکت و گلبولهای قرمز در برابر اشعه نسبتاً مقاومند و گمان میرود که سلولهای استرومال مغز استخوان نیز مقاوم باشند. تابش سطح بدن با بیشتر از Gy1/5< موجب پانسیتوپنی شدید در دو تا چهار هفته میشود. میزان LD50 اشعهی یونیزان حدود Gy4/5 است و تابش Gy10 یا بیشتر آن، صددرصد موجب مرگومیر میشود. نارسایی مغز استخوان به دوزاژ اشعه و کل مقدار دریافتی آن بستگی دارد.
کمخونی اپلاستیک ناشی از ازدیاد حساسیت دارویی (hypersensitive)
فهرست طویلی از داروها، از قبیل داروهای ضد تشنج، داروهای ضد التهاب غیراستروئیدی، آنتیبیوتیکها، داروهای ضد تکیاخته و ضد پرکاری تیروئید و ترکیبات طلا وجود دارند که ممکن است مصرف آنها در برخی افراد بدون عوارض و در برخی دیگر، موجب بروز کمخونی اپلاستیک شود که به آن پدیدهی ایدیوسینکراتیک گویند. ایجاد اپلازی با برخی داروها به مقدار مصرفی بستگی دارد. کلرامفنیکل ممکن است با هر دو سازوکار یادشده منجر به کمخونی اپلاستیک شود. یادآوری میشود که این دارو نیز با آسیب به DNA میتوکندری ممکن است موجب کمخونی سیدروبلاستیک شود. مدت زمان تأخیری بین مصرف دارو تا بروز کمخونی ممکن است بین دو تا سه ماه باشد.
افزایش آهن سرم و کاهش تعداد رتیکولوسیت از نشانههای صدمه به بافت خونساز، ناشی از مصرف داروهاست. آسیب به سلولهای سری اریتروئیدی موجب پسزدگی و بازگشت آهن از مغز استخوان به خون محیطی میشود و از این رو، افزایش آهن و کاهش رتیکولوسیتها را به دنبال دارد.
پدیدههای ایمونولوژیک در کمخونی آپلاستیک
نظریه رایج در کمخونی آپلاستیک اکتسابی بر این است که یک ویروس یا دارو با ایجاد واکنش ایمنی ناهنجار موجب تکثیر و فعال شدن الیگوکلونال سلولهای T-Cytotoxic میشود. پیوند سلولهای مادر خونساز و درمان با سرکوبگرهای ایمنی در کاهش اثرات تخریبی T-Cytotoxic گرچه مؤثر است ولی عود رخ میدهد و کلون غیرطبیعی سلول مادر ممکن است مسیر تبدیل به سندرم میلودیس پلاستیک و یا لوسمی حاد مایلوبلاستیک AML/MDS را طی کند.
شواهد خود ایمنی (Auto immunity) در ایجاد کمخونی آپلاستیک:
- بهبودی هماتولوژیکی با آنتی تیموسیت گلوبولین (ATG) و سیکلوسپورین در بیشتر بیماران
- فعال شدن CD8+ T خودواکنشی که موجب افزایش اینترفرون گاما و TNFα که نقش سرکوب بافت خونساز را دارد
- افزایش بیان FAS بر سطح سلولهای +CD34
- افزایش بیان فاکتورهای نسخهبرداری T-bet که با پیوند به پروموتور اینترفرون گاما موجب بیان بیشتر آن میشود
- آسیب به سلولهای تنظیمگر +CD4+،CD25+،FOXP که نتیجه آن ایجاد ریزمحیطی التهابی است.
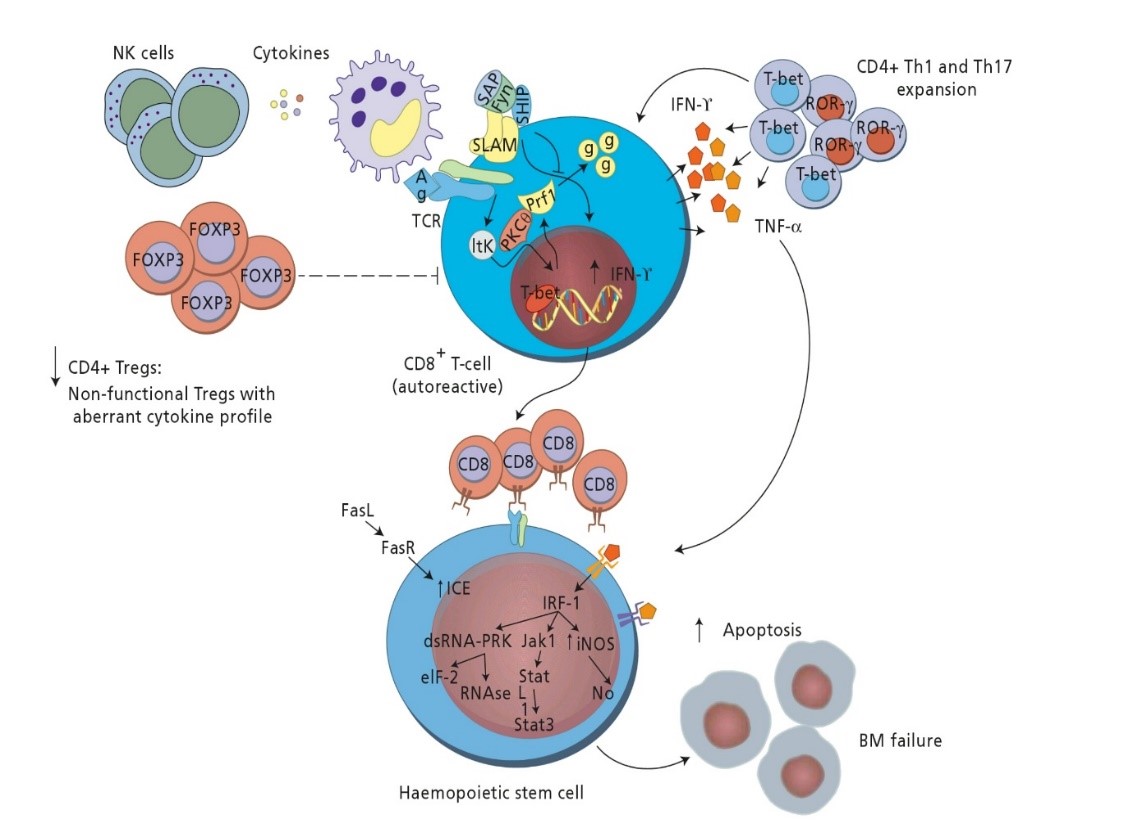
پدیدههای ایمونولوژیک با علل گوناگون، منجر به ترشح اینترفرون گاما و فاکتور نکروزدهندهی تومور میشود که بازدارندهی فعالیت بافت خونساز است. تهاجم سلولهای T سایتوتوکسیک به سلولهای خونساز و تولید انبوه اینترفرون گاما و فاکتور نکروزدهندهی بافتی که باعث بیان بیشتر Fas روی سلولهای خونساز بهمنظور آپوپتوز سلولهای +CD34 میشود، در اتیولوژی کمخونی نقش دارند
آزمایشهای تشخیصی در اولین مراجعه بیمار مشکوک به کمخونی آپلاستیک
- آزمایش CBC
پانسایتوپنی همراه با کاهش شدید رتیکولوسیت، نبود تغییرات دیسپلاستیک، نبود بلاست، نبود پلاکتهای درشت توپر، نبود گلبولهای قرمز پلیکروماژی و هستهدار، نبود طحال و کبد بزرگ و نبود غدد لنفاوی بزرگ در معاینه فیزیکی، شک را به سوی کمخونی آپلاستیک میبرد.
در کمخونی آپلاستیک، ماکروسیتوز و گرانولاسیون توکسیک شایع است. طحال بزرگ یا غدد لنفاوی بزرگ با نمای پانسایتوپنی ممکن است لوسمی سلولهای مودار را بهویژه وقتی که با کاهش شدید مونوسیت همراه باشد، مطرح کند. لوسمی و لنفوم هم ممکن است با نمای پانسایتوپنی و ارگانومگالی همراه باشند، غدد لنفاوی و طحال بزرگ در کمخونی اپلاستیک مشاهده نمیشود. گلبولهای قرمز در کمخونی اپلاستیک، نرموکروم تا ماکروسیت هستند، گرچه ممکن است در مغز استخوان تصویر مگالوبلاستوز مشاهده شود، سیدروبلاست حلقوی و تغییرات دیسپلاستیک هرگز در ردههای مایلوئیدی و مگاکاریوسیتی مشاهده نمیشود. تعداد سلولهای +CD34 در مغز استخوان اپلاستیک به علت تهاجم ایمونولوژیک کمتر از 0/3 درصد است؛ درحالیکه مقدار طبیعی آن 0/5 تا یک درصد است.
- آزمایشهای کبدی (Liver function tests)
گرچه در کمخونی آپلاستیک ناشی از هپاتیت سرولوژی ویروسی غالباً منفی است ولی ممکن است هنوز افزایش آنزیمهای کبدی مشاهده شود.
- اندازهگیری سطح ویتامین 12 B و اسید فولیک
کمخونی مگالوبلاستیک شدید با نمای پانسایتوپنی و ماکروسیتوز ظاهر میشود و ممکن است بتوان با گلبولهای شکسته، گلبولهای قرمز هستهدار مگالوبلاست و نوتروفیلهای هایپرسگمانته آن را از کمخونی آپلاستیک افتراق داد.
- آزمایشهای ویروسشناسی
آزمایشهای مربوط به انواع ویروسهای هپاتیت و آزمایش CMV بهویژه برای آن دسته از بیماران که کاندیدای پیوند سلولهای بنیادین خونساز هستند.
- آزمایشهای ANA و آنتیبادی علیه DNA دو رشتهای (dsDNA)
لوپوس و بیماریهای آتوایمیون دیگر علت غیرشایع کمخونی آپلاستیک هستند.
- رادیولوژی و سیتیاسکن
عکس سینه برای مشاهده عفونت یا فیبروز و تصویربرداری از شکم برای مشاهده آدنوپاتی و طحال و کبد بزرگ و کلیه غیرطبیعی در موارد مشکوک به کمخونی فانکونی.
- آزمایش آسپیره و بیوپسی مغز استخوان
گستره تهیهشده از آسپیراسیون مغز استخوان در غالب موارد رقیق (Diluted) و شبیه به خون محیطی است. نمای هایپوسلولاریتی با فقدان دیسپلازی در ردههای گرانولوسیتی و مگاکاریوسیتی مشاهده گردیده و مغز استخوان ممکن است لنفوسیتوز، پلاسماسل، ماکروفاژ و تعدادی ماست سل (Mast cell) را نشان دهد. افزایش سلولهای چربی، نمایی کندویی شکل به بیوپسی مغز استخوان داده و ممکن است گرههای کوچک خونساز در مغز استخوان هایپوسلولار مشاهده شود. رنگآمیزی برای سلولهای CD34+ ,(Immunostain) منفی یا تعداد بسیار کم را نشان میدهد.
پروفایل بیان ژن در سلولهای مادر خونساز در بیمار مبتلا به کمخونی آپلاستیک +AACD34 در بیش از 50 درصد موارد افزایش بیان ژنهای پاسخهای ایمنی برای تولید سایتوکاینها و آپوپتوز را نشان میدهد. آنالیز توالی نوکلئوتیدها با NGS (Next generation sequence) جهشهای سوماتیک در 20 تا 25 درصد بیماران مبتلا به کمخونی آپلاستیک بهویژه ژنهای 1 ASXL و DNMT3A که جهشهای بدخیمی مایلوئیدی هستند را نشان داده است. حضور این جهشها پیشبینی میکند که خطر تبدیل کمخونی آپلاستیک به AML/MDS در بیمارانی که بیش از 6 ماه از دریافت داروهای سرکوب گر ایمنی زنده ماندهاند، وجود دارد.
لیست آزمایشهای ضروری برای علتیابی کم خونی اپلاستیک

پاروویروس B19 که تنها میزبان آن انسان است، با تهاجم به گلبولهای قرمز هستهدار و پروژنیتورها (CFU-E) با پیشینهی کمخونی همولیتیک، موجب بروز کریز اپلاستیک یا بحران اپلاستیک میشود. گیرندهی ویروس، آنتیژن P است که روی سلولهای مادر خونساز وجود ندارد و روی گلبول قرمز و پروژنیتورها ظاهر میشود. افت بیش از 3 گرم هموگلوبین، کاهش شدید رتیکولوسیت و ناپدیدشدن مرفولوژی پلیکروماژی و گلبولهای قرمز هستهدار از خون محیطی در بیماری که کمخونی همولیتیک دارد، ممکن است نشانهی عفونت با پاروویروس B19 باشد. بهبودی از عفونت پاروویروس موجب روانه شدن انبوه گلبول قرمز هستهدار به خون محیطی به علت جبران مغز استخوان میشود.
ویروس HIV ممکن است با نوتروپنی و کاهش پلاکت همراه باشد. بروز ترومبوسیتوپنی امکان دارد اولین نشانهی ابتلا به ویروس باشد. مغز استخوان در عفونت با ویروس HIV ممکن است تصویری شبیه به سندرمهای میلودیسپلاستیک داشته باشد.
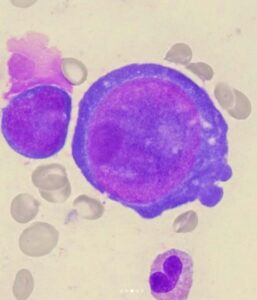
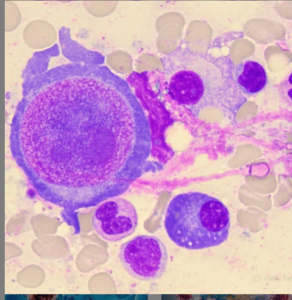
فقدان سری اریتروئیدی در مغز استخوان، ناشی از عفونت با پاروویروس B19؛
در بحران اپلاستیک بهجز تعداد محدودی پرونرموبلاست غولآسا با امکان مشاهده انکلوزیون هستهای، سلولهای دیگر اریتروئیدی مشاهده نمیشود
سندرم تلومر کوتاه (short telomere syndrome)
سندرم کوتاه شدن طول تلومر با علائم بالینی گوناگون در رابطه با طول تلومر همراه است. نارسایی مغز استخوان، فیبروز ریه، سیروز کبد، بیماریهای گوارشی و دیستروفی ناخن، لکوپلاک در دهان و مخاط و پیگمانه شدن غیرطبیعی پوست از علائم کوتاه شدن طول تلومر است البته تمام علائم در یک بیمار مشاهده نمیشود. علائم ممکن است در کودکی یا بزرگسالی شروع شود.

تلومرها توالی نوکلئوپروتئینهای ویژهای هستند که از صدها تا هزاران سکانس تکراری (گاهی تا 3000) پشت سر هم TTAGGG تشکیل شدهاند و در انتهای کروموزومها قرار میگیرند. تلومرها انتهای کروموزومها را از تخریب، تجزیه شدن، نوآرایی و الحاق انتهایی حفظ میکنند. تلومر به عنوان یک ساعت مولکولی یا ساعت میتوزی تعداد تقسیمات سلول را مشخص کرده و کوتاه شدن آن سیگنالهای آپوپتوز را در پی دارد. واحدهای پروتئینی شلترین (SHETRIN) به توالیهای تلومر پیوند داده و موجب حفظ و نگهداری ساختمان تلومر میگردد. برای جلوگیری از کوتاه شدن طول تلومر در طی همانندسازی، آنزیم تلومراز که یک ترنس کریپتاز معکوس است با اضافه کردن توالیTTAGGG موجب رشد آن میگردد.
پیشرفتهای اخیر پزشکی در زمینه ژنتیک منجر به شناسایی طیفی از بیماریهای ژنتیکی در رابطه با طول و ساختار تلومر شده است. گفتنی است که روش زندگی سالم، ورزش، آنتیاکسیدانها و استاتینها نقش مهمی در فعال بودن تلومراز دارند، در حالی که سیگار، افزایش فشار خون، دیابت، چاقی، الکل و به ویژه استرسهای اکسیداتیو و روانی با کوتاه شدن طول تلومر همراهی دارند. اختلالات تلومر با عوارضی از قبیل سختی عروق کرونر، نارسایی قلب، سکته و پیری زودرس و دستهای از کمخونیها در رابطه است.
از مهمترین ژنهای جهشیافته در سندروم تلومر کوتاه TERT و TERCمیباشند. دیسکراتوز ارثی یکی از بیماریهای خانواده تلومر کوتاه است که با کمخونی آپلاستیک بروز میکند، از این رو معاینه بیمار مبتلا به کمخونی آپلاستیک یا آگاهی از سابقه فیبروز ریه و کبد و مشاهدات غیرطبیعی در ناخن و پوست و دهان در بیمار یا اقوام درجه اول در تشخیص علت کمخونی آپلاستیک مهم است. اندازهگیری طول تلومر با روشهای Fish Flow و یا qPCR Multiplex امکانپذیر است.
آزمایش کلاستوژن یا شکستگیهای متعدد کروموزومی با مجاورت سلولهای لنفوسیت با دای اپوکسی بوتان (DEB) و یا میتومایسین (MMC) برای تشخیص کمخونی فانکونی به کار میرود. این کمخونی بهصورت آپلاستیک در سنین 5 تا 7 سالگی بروز کرده و با اختلالات اسکلتی همراه است و با توجه به اینکه کمخونی حتی تا 40 سالگی هم گزارش شده است، از این رو بایستی برای بیمارانی که کاندیدای دریافت پیوند هستند مورد بررسی قرار گیرند.
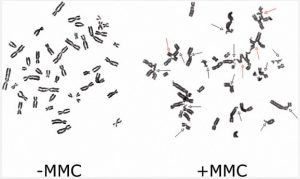
تشخیص کمخونی فانکونی با کشت لنفوسیتهای بیمار در مرحلهی متافاز در حضور DEB (دای اپوکسی بوتان) یا میتومایسین صورت میگیرد و حداقل پنجاه سلول متافاز برای تشخیص شکستگی کروموزومی، بهویژه شکستگیهای شعاعی بررسی میشوند و نتایج با کنترل نرمال مقایسه میگردد. نتایج ناهنجاری با تعداد سلولها با شکستگی کروموزومی و شکلهای شعاعی گزارش میشود. شکستگیهای متعدد و فیوژن کروموزومها در مجاورت میتومایسین در کمخونی فانکونی در شکل بالا مشاهده میشود؛ تعداد شکستگیها با میانگین 6/69 به ازای هر سلول در مقابل 0/06 برای هر سلول نرمال است.
کمخونی فانکونی سندرم ناپایداری کروموزمی است که با اختلالات ارثی و پانسیتوپنی تدریجی و آسیبپذیر به بدخیمیهای خونی و بافت توپر بروز میکند. بیماران فانکونی، بسیار در معرض خطر ابتلا به سندرمهای مایلودیسپلاستیک و لوسمیهای مایلوبلاستیک هستند؛ علاوه بر این تومورهای بافت توپر، بهویژه سرطانهای سر و گردن، پوست، گوارش و سیستم ادراری نیز شایع است. آزمایش CBC در بدو تولد نرمال است؛ ولی پانسیتوپنی بهتدریج و با میانگین سنی هفت سال رخ میدهد و بهندرت ممکن است در دوران بلوغ مشاهده شود. آزمایش طلایی کمخونی، مشاهدهی شکستگیهای متعدد کروموزومی در سلولهای بیمار در مجاورت با مواد آسیبزای DNA، از قبیل میتومایسین و دای اپوکسی بوتان است.
برخی اختلالات ارثی در کمخونی فانکونی عبارتاند از:
ـ آنومالی اسکلتی، مانند اختلال در شکلگیری انگشت شست و استخوان رادیوس و اسکولیوز
ـ قد کوتاه
ـ لکههای شیرقهوهای یا لکههای بیرنگ روی پوست
ـ آنومالی کلیه، مانند کلیهی نعل اسبی و گونادهای شکلنگرفته
ـ اختلال گوارشی و قلبی و عصبی
ـ هیدروسفال
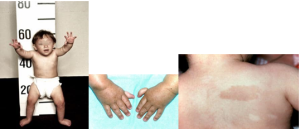
برخی اختلالات ارثی در کمخونی فانکونی
آنالیز NGS (آنالیز توالی ژن) برای جهشهای سوماتیک در افتراق کمخونی آپلاستیک از مایلو دیسپلاستیک هایپوسلولار سفارش میشود. عفونتهای سل ممکن است با پانسایتوپنی و شبیه آنمی آپلاستیک ظاهر شود که در این حالت مشاهده گرانولوما در مغز استخوان افتراقدهنده است. در نوتروپنی و پانسایتوپنی مربوط به بیاشتهایی عصبی (Anorexia nervosa)، ورم سلولهای استرومال، تبــــــدیل ژلاتینی و آتروفی سروز (Serous atrophy) با رسوب ماده پروتئینی شکل در مغز استخوان دیده میشود. حالت فوق در الکلیسم و نارسایی مزمن قلبی و گرسنگی هم گزارش شده است.
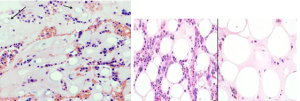
مغز استخوان هایپوسلولار با تبدیل ژلاتینی و آتروفی سروز (Serous atrophy) با رسوب ماده پروتئینی شکل در مغز استخوان
تمام بیماران مبتلا به کمخونی آپلاستیک بایستی برای هموگلوبیناوری حمله شبانه (PNH) مورد بررسی قرار گیرند. PNH، به علت جهش سوماتیک در ژن PIGA روی کروموزوم X است. جهش، موجب حساس شدن و همولیز گلبولهای قرمز در برابر تهاجم سیستم کمپلمان میشود. فراوردهی ژن PIGA برای اتصال CD55 و CD59 به گلبولهای قرمز و محافظت از آنها در برابر هجوم کمپلمان ضروری است. تعداد کمی سلول PNH در بیماران مبتلا به کمخونی اپلاستیک وجود دارد. در برخی بیماران مبتلا به کمخونی اپلاستیک، افزایش سلولهای PNH موجب بروز علائم بالینی و کمخونی PNH میشود. با تعیین سایز کلون PNH میتوان کمخونی همولیتیک خالص PNH با تصویر آپلاستیک و نارسایی مغز استخوان را از کمخونی آپلاستیک همراه با PNH (AA/PNH) افتراق داد.
کلون هموگلوبیناوری شبانه PNH (PNH clone) تا 50 درصد موارد کمخونی آپلاستیک و با نسبت کمتری در سندرمهای میلودیس پلاستیک گزارش گردیده است. چگونگی شکل گرفتن گلبولهای قرمز PNH در کمخونی آپلاستیک مورد پرسش است. با توجه به اینکه کمخونی آپلاستیک یک بیماری آتوایمیون با تهاجم سلولهای +TCD8 به بافت خونساز است، گمان میرود که در سلول مادر خونساز در کمخونی آپلاستیک در لنــگرگاه (GPI (Glycosyl phosphatidyl inositol تغییراتی حاصل شود که با فقدان GPI از تهاجم ایمنی فرار کند و موجب توسعه کلون PNH گردد. شناسایی کلون PNH در بیماران با کمخونی همولیتیک، نارسایی مغز استخوان و ترومبوز سفارش میشود.
برای شناسایی کلون PNH میتوان از آزمایشهای فلورسانس FLAER، نبود مارکرهای 55CD و 59CD بر سطح گلبولهای قرمز، گرانولوسیتها و مونوسیتها با فلوسایتومتری استفاده کرد.
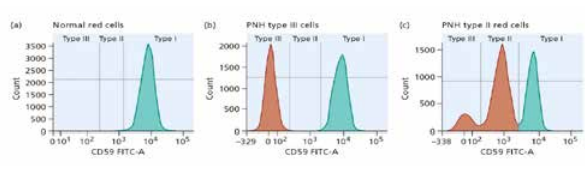
حضور CD55 و CD59 بر سطح سلول با آنتیبادی منوکلونال نشاندار با روش فلوسایتومتری آنالیز میشود. گلبولهای PNH فاقد این دو پروتئین هستند. مارکرهای CD55 و CD59 عهدهدار حفاظت گلبول قرمز در مقابل تهاجم سیستم کمپلمان هستند که از طریق GPI به غشای گلبول وصل میشود. در تست Flear سم ارولیزین نشاندار، مستقیماً به ترکیب گلیکان از اجزای پروتئین GPI متصل میشود. گلبولهای PNH فاقد GPI هستند
مدیریت کمخونی آپلاستیک
با توجه به این که بیماران مبتلا به کمخونی آپلاستیک بهطور مزمن نیاز به تزریق خون و فراورده پلاکت دارند، توجه به نکات زیر ضروری است:
- برای کم شدن شانس آلوایمونوزاسیون و ساخته شدن آنتیبادی علیه HLA از فراورده LR-RBC استفاده کنید که 99/9 درصد از گلبولهای سفید کیسه توسط لکوتراپ برداشت شده است. این فراورده نه تنها ساختن آنتیبادی علیه آنتیژنهای HLA را کاهش میدهد، بلکه از سرایت ویروس CMV هم تا حد زیادی جلوگیری کرده و به علاوه واکنشهای تب و لرز را در بیمار کاهش میدهد.
- چنانچه بیمار علیه آنتیژنهای HLA تحریک شود، شانس ایجاد پدیده رفراکتوری در تزریق پلاکت وجود دارد؛ بدین مفهوم که بیمار به تزریق پلاکت پاسخ نداده و تزریق پلاکت موجب افزایش پلاکت و بند آمدن خونریزی نمیشود.
- برای بیمار مبتلا به کمخونی آپلاستیک که داروی ATG میگیرد در موقع نیاز به خون بایستی از فراورده Irradiated و LR-RBC استفاده شود؛ بدین مفهوم که خون لکوتراب شده در معرض تابش اشعه X یا 2500 راد اشعه گاما قرار گیرد تا لنفوسیتهای کیسه غیرفعال شده و موجب سندرم گرافت که مرگ را در پی دارد نشود. سندرم گرافت با تهاجم لنفوسیتهای زنده کیسه به ارگانهای حیاتی و با پدیدار شدن بثورات جلدی و تب آغاز شده و به کمخونی آپلاستیک مرگبار منتهی میشود.
- سطح پلاکت بیشتر از 10000 در بیماری که تب و عفونت ندارد کافی است ولی چنانچه تب و عفونت دارد بایستی بیشتر از 20000 باشد. چنانچه بیمار خونریزی دارد بایستی به بیشتر از 50000 افزایش یابد. اگر بیمار با ساختن آنتیبادی به تزریق پلاکت، رفراکتوری باشد بایستی اهداکننده پلاکت را از میان اقوام درجه اول با تشابه HLA-A و HLA-B انتخاب کرد و فراورده پلاکتی آفرز را پس از اشعه دادن به بیمار تزریق کرد.
روشهای انتخابی برای درمان کمخونی آپلاستیک، پیوند آلوژنیک سلولهای بنیادی خونساز (HSC) و یا سرکوبگرهای ایمنی با آنتی تیموسیت گلوبین (ATG) و سایکلوسپورین (CSA) است.
انتخاب روش درمان بستگی به سن بیمار، شدت بیماری و در دسترس بودن برادر یا خواهر با تطابق HLA دارد. در بیماران با کمخونی شدید و سن زیر 50-35 سالگی اولین خط درمان HSCT (پیوند سلول مادر خونساز) از دهنده با شباهت HLA است. آنالیز دادهها نتایج مشابهی در پیوند بین سنهای 30 تا 40 و 40 تا 50 داده است. برای تمام بیمارانی که نیاز به درمان دارند به ویژه کمخونی غیر شدید یا با سن بالاتر از 50 سال، تجویز سرکوبگرهای ایمنی اولین انتخاب است و چنانچه پاسخی با اولین دوزاژ درمانی نداد، پیوند سلولهای بنیادین از دهنده غیر وابسته به شرطی که مطابقتدار در دسترس نباشد، انجام میشود.
آنتیتیموسیت گلوبین (ATG) پلیکلونال IgG است که با تحریک ایمنی اسب یا خرگوش و با تزریق تیموسیتهای انسان تهیه میشود. سازوکار ATG تخلیه سلولهای T و لیز آنها با واسطه کمپلمان، از بین بردن سلولهای +TCD8 فعال با بیان FAS و آپوپتوز و اثرات سایتوتوکسیسیته با واسطه آنتیبادی (ADCC) و تحریک مستقیم T رگولاتوری است. آنتیتیموسیت گلوبین از منبع خرگوش بیشتر از منبع اسب خاصیت سرکوب ایمنی دارد و طول زمان لنفوپنی بیشتر بوده و نیز افینیتی بالا برای لنفوسیتهای انسان دارد ولی با این حال ATG تهیه شده از اسب بر ATG خرگوش ترجیح داده میشود و میزان پاسخ 68 درصد در 6 ماه در مقایسه با 37 درصد نسبت به ATG تهیه شده از خرگوش است.

شواهدی از خودایمنی در بروز کمخونی اپلاستیک در دست است؛ شاید بهترین شاهد بر سازوکارهای ایمنی در ایجاد کمخونی اپلاستیک، تجویز ATG به بیماران و پاسخ مثبت و بهبودی در حدود دو سوم از مبتلایان باشد. پدیدههای ایمونولوژیک با علل گوناگون، منجر به ترشح اینترفرون گاما و فاکتور نکروزدهندهی تومور میشود که بازدارندهی فعالیت بافت خونساز است
برای جلوگیری از بیماری سرم (Serum sickness) که با آرترالژی، میالژی، راش، تب و پروتئیناوری ظاهر میگردد، از استروئیدها حین درمان استفاده میشود. بهترین پاسخ ATG در بیماران با سن کم، رتیکولوسیت بیشتر از 25000 در میلیمتر مکعب، شمارش مطلق لنفوسیتی بیشتر از 1000 و کلون PNH کمتر از 0/003 درصد مشاهده میگردد. ممکن است برای بیمارانی که با ATG با منبع اسب پاسخ ندادهاند از یک دوره درمان با ATG تهیه شده از خرگوش یا تجویز Alemtuzumab (Anti-CD52) استفاده شود.
افزایش طول عمر بیماران به دنبال درمان با ATG خطراتی از قبیل اختلالات کلونال سلول مادر از قبیل PNH و MDS و یا AML به دنبال دارد.
علت اینکه چرا تمام بیماران به درمان سرکوبگرهای ایمنی پاسخ نمیدهند میتوان به تهاجم شدید سیستم ایمنی و تخلیه مغز استخوان از سلول مادر، دوزاژ کم دارو و تلومر کوتاه اشاره کرد. برخی از بیماران برای جلوگیری از عود کمخونی، به سیکلوسپورین برای طولانی مدت وابسته میگردند. برای موفقیتآمیز بودن پیوند سلولهای بنیادی خونساز احتیاج به تزریق حداقل 8 10×3 از سلولهای هستهدار مغز استخوان به ازای هر کیلوگرم وزن بیمار و یا 6 10×2 از سلولهای +CD34 به ازای هر کیلوگرم است. سلولهای مادر تهیه شده از خون محیطی شانس واکنشهای سندرم گرافت را بالا میبرد.
برای افزایش هموگلوبین، استفاده از خون با کاهش گلبولهای سفید (LR-RBC) و خون اشعهدیده شانس گرفتن پیوند را بیشتر میکند. برای جلوگیری از تحریک آنتیژنی علیه آنتیژنهای ماینور (Minor HLA) سفارش میشود از فراوردههای خونی اهداکننده قبل از پیوند استفاده نگردد. سلول مادر خونساز فاقد آنتیژنهای ABO است و ناسازگاری ABO موجب رد حاد پیوند نمیگردد. تزریق خون مکرر موجب انباشت آهن و هموسیدروز میگردد که دارای اثرات نامطلوب روی پیوند است. چنانچه سطح فریتین بیشتر از 1000 میکروگرم در لیتر گردد، استفاده از چلاتورهای آهن اجتنابناپذیر است. از چلاتور دفیریپرون (Deferiprone) به علت اینکه با شیوع زیاد آگرانولوسیتوز همراه است در کمخونی آپلاستیک استفاده نمیشود.
https://www.mayoclinic.org/diseases-conditions/aplastic-anemia/symptoms-causes/syc-20355015
https://medlabnews.ir/%da%a9%d9%85%e2%80%8c%d8%ae%d9%88%d9%86%db%8cmcv-%d9%88-rpi/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام