متابولیسم کلسترول و اختلالات آن
مراد رستمي: کارشناس ارشد بیوشیمی بالینی، دانشگاه علوم پزشكي جندیشاپور اهواز
معصومه جرفی: کارشناس ارشد میکروبشناسی، دانشگاه علوم پزشكي جندیشاپور اهواز
مقدمه:
کلسترول در بافتها و در پلاسما به شکل کلسترول آزاد یا به شکل ذخیرهای آن، متصل به اسید چرب با زنجیره بلند بهصورت کلسترول استر وجود دارد. در پلاسما، هر دو فرم در لیپوپروتئینها منتقل میشوند. کلسترول یک لیپید آمفیپاتیک بوده و یک جزء ساختمانی ضروری غشاها است که برای حفظ نفوذپذیری و سیالیت آنها مهم است و لایه خارجی لیپوپروتئینهای پلاسماست. این ماده در بافتهای مختلف از استیل کوآ ساخته میشود و پیش ساز تمامی استروئیدها شامل کورتیکواستروئیدها، هورمونهای جنسی، اسیدهای صفراوی و ویتامین D است.
کلسترول به دلیل اینکه یک محصول متابولیکی در حیوانات است، در غذاهای حیوانی مثل زردهی تخممرغ، گوشت، جگر و مغز یافت میشود. LDL پلاسما، وسیله تأمین کلسترول و استر کلسترول برای بافتهای مختلف است. کلسترول آزاد بهوسیله HDL پلاسما در طی فرآیندی به نام انتقال معکوس کلسترول، از بافتها به کبد منتقلشده و در آنجا، یا بهصورت کلسترول آزاد و یا بعد از تبدیلشدن به اسیدهای صفراوی، از بدن دفع میگردد. کلسترول، یک جزء اصلی سنگهای صفراوی است. همچنین نقش اصلی آن در فرآیندهای پاتولوژیک، عمل کردن بهعنوان فاکتوری است که موجب آترواسکلروز شریانهای حیاتی و درنتیجه، بیماری عروق مغزی، قلبی و محیطی میشود.
کمی بیش از نصف کلسترول بدن از طریق بیوسنتز (حدود 700 میلیگرم در روز) و بقیه از طریق رژیم غذایی معمول، تأمین میشود. کبد و روده، هرکدام حدود 10% از کل کلسترول سنتز شده در انسان را میسازند. تمامی بافتهای دارای سلولهای هستهدار، قادر به سنتز کلسترول میباشند. محل سنتز کلسترول، در شبکه آندوپلاسمی و بخشهای سیتوزولی میباشد.
بیوسنتز کلسترول
بیوسنتز کلسترول را میتوان به 5 مرحله تقسیم نمود:
مرحله 1- سنتز موالونات از استیل کوآ:
3- هیدروکسی-3- متیل گلوتاریل کوآ (HMG-CoA)، بهوسیله واکنشهایی که جهت تولید اجسام کتونی در میتوکندری مورد استفاده قرار میگیرند، تولید میگردد. سنتز کلسترول، در خارج از میتوکندری انجام شده و از این نظر، با سنتز اجسام کتونی متفاوت هستند. ابتدا 2 ملکول استیل کوآ در طی یک واکنش سیتوزولی که توسط تیولاز کاتالیز میشود، باهم ترکیب شده و تولید استواستیل کوآ را مینمایند. استواستیل کوآ به کمک HMG-CoA سنتاز به یک ملکول دیگر استیل کوآ ترکیب شده و تولید
HMG-CoA مینماید. HMG-CoA توسط HMG-CoA ردوکتاز و با استفاده از NADPH به موالونات احیا میگردد. این واکنش که توسط HMG-CoA ردوکتاز کاتالیز میگردد، مرحله تنظیمی اصلی مسیر سنتز کلسترول و محل اثر مؤثرترین داروهای کاهنده کلسترول (مانند استاتینها) میباشد.
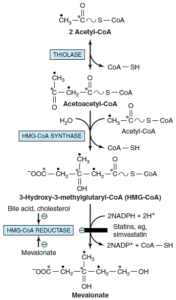
مرحله 2- تشکیل واحدهای ایزوپرنوئید از موالونات با از دست دادن CO2:
موالونات بهطور متوالی توسط 3 کیناز و با استفاده از ATP فسفریله شده و بعد از دکربوکسیله شدن، واحد ایزوپرنوئیدی فعال یعنی ایزوپنتیلدیفسفات را ایجاد میکند.
مرحله 3- ترکیب 6 واحد ایزوپرنوئید و تشکیل اسکوالن:
ایزوپنتنیلدیفسفات با تغییر پیوند دوگانه به ایزومر خود یعنی دی متیلآلیلدیفسفات تبدیل میشود. سپس دیمتیلآلیلدیفسفات با یک ملکول دیگر ایزوپنتنیلدیفسفات ترکیب شده و واسطه ده کربنی ژرانیل دی ففسات را به وجود میآورد. این ماده با یک ایزوپنتنیلدیفسفات دیگر ترکیب شده و فارنسیل دی فسفات را حاصل میکند. دو ملکول فارنسیل دی فسفات از انتهای دی فسفات باهم ترکیب شده و اسکوالن را تشکیل میدهند. در این واکنش، ابتدا پیروفسفات معدنی حذف میشود و پره اسکوالن دی فسفات حاصل میشود، سپس با احیا شدن بهوسیله NADPH، ملکول پیروفسفات معدنی دیگر نیز حذف میشود.
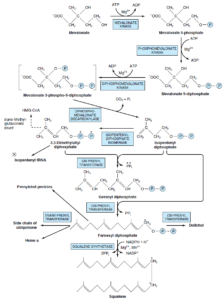
مرحله 4- حلقوی شدن اسکوالن و ایجاد استروئید مادر لانوسترول:
اسکوالن میتواند پیچ بخورد و ساختاری کاملاً شبیه هسته استروئید را ایجاد نماید. قبل از اینکه حلقه بسته شود، اسکوالن بهوسیله یک اکسیداز چندکاره در شبکه اندوپلاسمی به نام اسکوالن اوپسیداز به اسکوالن 2 و 3- اپوکسید تبدیل میشود. هنگام حلقوی شدن که توسط آنزیم اکسیدواسکوالنه لانوسترول سیکلاز کاتالیز میشود، گروه متیل متصل به C14 به C13 و گروه متصل به C8 به C14 منتقل میشود.
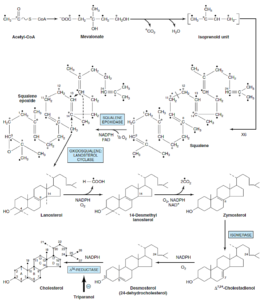
مرحله 5- ایجاد کلسترول از لانوسترول:
تولید کلسترول از لانوسترول در غشاهای شبکه اندوپلاسمی اتفاق میافتد و شامل تغییراتی در هسته استروئید و زنجیره جانبی است. گروههای متیل متصل به C14 به C4 برداشته میشوند و بدین ترتیب 14- دس متیل لانوسترول و سپس زایموسترول تولید میشود. سپس پیوند دوگانه در موقعیت C8-C9 در دو مرحله به موقعیت C5-C6 منتقل و دسموسترول تشکیل میشود. درنهایت، پیوند دوگانه زنجیره جانبی، احیا شده و کلسترول حاصل میشود.
تنظیم سنتز کلسترول:
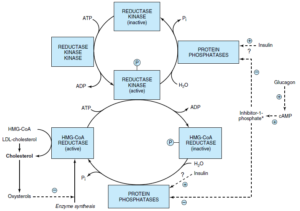
تنظیم سنتز کلسترول در مرحله HMG-CoA ردوکتاز صورت میگیرد. HMG-CoA ردوکتاز در کبد بهوسیله موالونات (محصول واسطه واکنش) و کلسترول (محصول اصلی مسیر) مهار میشود. کلسترول و متابولیتهای آن با فعال کردن یک فاکتور رونویسی به نام پروتئین متصل شونده به قسمت تنظیمی استرول (Sterol Regulatory Element-Binding Protein; SREBP)، رونویسی
HMG-CoA ردوکتاز را مهار میکنند. انسولین و هورمونهای تیروئیدی، فعالیت HMG-CoA ردوکتاز را افزایش و گلوکاگن و گلوکوکورتیکوئیدها، فعالیت آن را کاهش میدهند.
تعادل کلسترول در بافتها:
افزایش کلسترول سلول درنتیجه برداشت لیپوپروتئینهای حاوی کلسترول بهوسیله گیرندهها مثل گیرنده LDL یا Scavenger receptor، برداشت کلسترول آزاد از لیپوپروتئینهای غنی از کلسترول به غشای سلولی، سنتز کلسترول و هیدرولیز استرهای کلسترول بهوسیله آنزیم کلستریل استر هیدرولاز اتفاق میافتد. کاهش کلسترول سلول به علت خروج کلسترول از غشا به HDL از طریق ABCA-1، ABCG-1 یا SR-B1، استریفیه شدن کلسترول بهوسیله آنزیم ʺآسیل کوآ کلسترول آسیل ترانسفرازʺ (Acyl-CoA: cholesterol acyltransferase) و مصرف کلسترول برای سنتز استروئیدهای دیگر نظیر هورمونها یا اسیدهای صفراوی در کبد رخ میدهد.
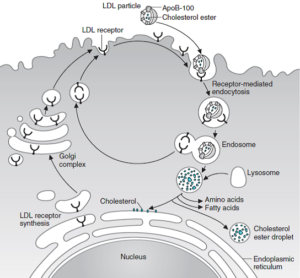
تنظیم گیرنده LDL:
گیرندههای LDL در حفراتی واقع در سطح سلول که توسط پروتئینی به نام کلاترین پوشیده شدهاند، قرار دارند. در انتهای آمینی این گیرندهها که از غشا بیرون زده است، ناحیه متصل شونده به B100 قرار دارد. LDL پس از اتصال به گیرنده، به شکل دستنخورده از طریق اندوسیتوز برداشت میشود. آپوپروتئین و استر کلسترول در لیزوزومها هیدرولیز شده و کلسترول به درون سلول انتقال داده میشود. گیرندهها مجدداً به سطح سلول برمیگردند. این ورود کلسترول موجب مهار رونویسی ژنهای کدکننده HMG-CoA ردوکتاز و آنزیمهای دیگر دخیل در سنتز کلسترول و نیز خود گیرنده LDL از طریق مسیر SREBP میشود و بنابراین، سنتز و برداشت کلسترول بهطور هماهنگی مهار میشود.
همچنین فعالیت ACAT تحریک شده و کلسترول استریفیه میشود. در این فرآیند، فعالیت گیرنده LDL در سطح سلول بهوسیله کلسترول موردنیاز برای سنتز غشا، هورمونهای استروئیدی و یا اسیدهای صفراوی تنظیم میشود.
![]()
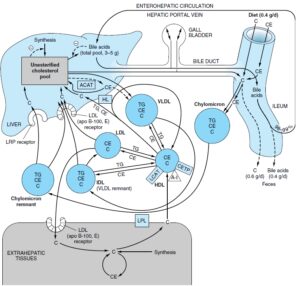
انتقال کلسترول
کلسترول در پلاسما در لیپوپروتئینها انتقال مییابد که بخش اعظم آن، استر کلسترول است و در انسانها، بیشترین نسبت آن در LDL وجود دارد. کلسترول رژیم غذایی در عرض چند روز با کلسترول پلاسما و در عرض چند هفته با کلسترول بافتی به تعادل میرسد. استر کلسترول رژیم غذایی به کلسترول هیدرولیز میشود و سپس همراه با کلسترول غیراستریفیه و سایر لیپیدهای غذایی بهوسیله روده جذب میشود. این کلسترول همراه با کلسترول سنتز شده در روده وارد ساختمان شیلومیکرونها میشود. 90- 80 درصد کلسترول جذب شده، در مخاط روده با اسیدهای چرب دارای زنجیره بلند استریفیه میشود. 95
درصد کلسترول شیلومیکرون بهصورت باقیماندههای شیلومیکرون به کبد تحویل داده میشود و قسمت اعظم کلسترول ترشحشده از کبد بهصورت VLDL، در جریان تشکیل IDL و درنهایت LDL، حفظ میشود. این کلسترول بهوسیله گیرنده LDL در کبد و بافتهای غیرکبدی برداشت میشود.
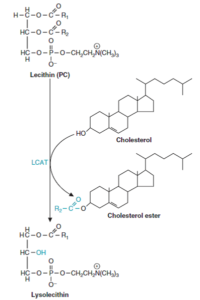
عملکرد LCAT:
فعالیت لسیتین کلسترول آسیل ترانسفراز (Lecithin cholesterol acyltransferase; LCAT) با HDL حاوی آپو A-I مرتبط است. با استریفیه شدن کلسترول در HDL، یک گرادیان غلظتی به وجود میآید و کلسترول را از بافتها و لیپوپروتئینهای دیگر به داخل HDL میکشاند و با این ترتیب، HDL را قادر میسازد که در انتقال معکوس کلسترول شرکت نماید. LCAT، اسید چرب موردنیاز برای استریفیه کردن کلسترول را از ملکول لسیتین تأمین مینماید.
عملکرد ACAT:
فعالیت آسیل کوآ کلسترول آسیل ترانسفراز (Acyl-CoA:cholesterol acyltransferase; ACAT)، استریفیه کردن کلسترول در داخل سلولها میباشد. ACAT برای استریفیه کردن کلسترول، از آسیل کوآ (اسیدهای چرب فعال آزاد) استفاده میکند.
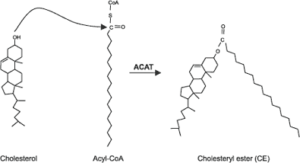
عملکرد پروتئین ناقل استر کلستریل:
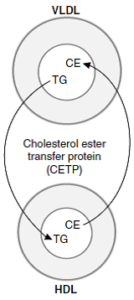
پروتئین ناقل استر کلستریل که با HDL مرتبط است، در پلاسمای انسان و بسیاری از گونههای دیگر یافت میشود. این پروتئین، انتقال استر کلستریل از HDL به VLDL، IDL و LDL را تسهیل میکند. در این فرآیند، استر کلستریل با تری آسیل گلیسرول معاوضه میشود و بدین ترتیب موجب کاهش مهار LCAT بهوسیله محصول آن در HDL میشود؛ بنابراین در انسان، قسمت اعظم استر کلستریل تولید شده توسط LCAT، از طریق باقیمانده (IDL) VLDL یا LDL به کبد راه مییابد. HDL2 غنی از تریآسیلگلیسرول، کلسترول خود را در چرخه HDL به کبد تحویل میدهد.
دفع کلسترول
کلسترول به فرم غیراستریفیه و یا پس از تبدیل شدن به اسیدهای صفراوی در کبد، از طریق صفرا در بدن دفع میشود.
کوپروستانول، استرول اصلی مدفوع است و قسمتی از کلسترول، از این طریق دفع میگردد. این استرول در قسمتهای انتهایی روده بهوسیله باکتریها، از طریق مسیر مستقیم و یا غیرمستقیم از کلسترول تولید میشود.
![]()
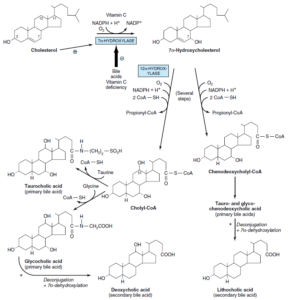
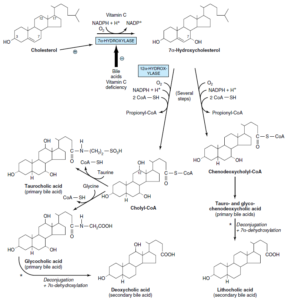
تولید اسیدهای صفراوی از کلسترول
اسیدهای صفراوی اولیه:
در کبد و از کلسترول تولید میشوند. اسیدهای صفراوی اولیه شامل اسید کولیک (بیشترین میزان را دارد) و اسید کنوداکسی کولیک میباشند. آنزیم 7 آلفا- هیدروکسیلاز (میکروزومی)، اصلیترین مرحله تنظیمی در بیوسنتز اسیدهای صفراوی میباشد. این آنزیم یک منواکسیژناز بوده و برای فعالیت خود به O2، NADPH و سیتوکروم P450 نیاز دارد.
مسیر بیوسنتز اسیدهای صفراوی به 2 مسیر فرعی تقسیم میشود:
1- مسیر کولیل کوآ (حاوی یک گروه آلفا- هیدروکسیل اضافی در موقعیت 12)
2- مسیر کنوداکسی کولیل کوآ
اسیدهای صفراوی اولیه با گلیسین و تورین و در پراکسی زومهای کبدی، کنژوگه شده و سپس وارد صفرا میگردند. نسبت کنژوگههای گلیسین به کنژوگههای تورین در انسان، بهطور طبیعی 3 به 1 میباشد. اسیدهای صفراوی در صفرای قلیایی (pH 7.6-8.4) به شکل نمک وجود دارند.
اسیدهای صفراوی ثانویه:
تولید اسیدهای صفراوی ثانویه از اسیدهای صفراوی اولیه، در روده و توسط باکتریهای آن و از طریق دکنژوگاسیون و 7 آلفا- دهیدروکسیلاسیون صورت میگیرد. اسیدهای صفراوی ثانویه شامل اسید داکسی کولیک و اسید لیتوکولیک میباشند.
دفع و گردش رودهای- کبدی اسیدهای صفراوی
جذب محصولات هضم چربی ازجمله کلسترول، در 100 سانتیمتر ابتدایی روده کوچک صورت میگیرد. جذب اسیدهای صفراوی اولیه و ثانویه در ایلئوم انجام میشود. 99- 98% اسیدهای صفراوی از طریق گردش خون باب به کبد (گردش رودهای- کبدی) برمیگردند. جذب اسید لیتوکولیک (به دلیل نامحلول بودن)، بهطور قابلملاحظهای، کمتر میباشد. بخش کوچکی از نمکهای صفراوی، جذب نشده و از طریق مدفوع دفع میگردند که همین بخش کوچک، یک مسیر اصلی برای دفع کلسترول است. روزانه، ذخیره اسیدهای صفراوی (حدود 5- 3 گرم)، 10- 6 بار از چرخه رودهای عبور میکنند. میزان ساختهشدن روزانه اسیدهای صفراوی، معادل میزان دفع شدهی آنها در مدفوع میباشد.
تنظیم سنتز اسیدهای صفراوی
مرحله اصلی محدود کننده سنتز اسیدهای صفراوی، واکنش کلسترول 7 آلفا- هیدروکسیلاز میباشد. تنظیم پسنورد این آنزیم، توسط گیرنده هستهای متصل شونده به اسید صفراوی ʺ گیرنده فارنسوئید ایکس (Farnesoid X receptor; FXR)ʺ صورت میگیرد. فعال شدن FXR، متعاقب افزایش اسیدهای صفراوی در گردش رودهای- کبدی و مهار رونویسی ژن کلسترول 7 آلفا- هیدروکسیلاز صورت میگیرد. اهمیت ویژه کنوداکسی کولیک اسید در فعال کردن FXR میباشد.
عوامل مؤثر در میزان کلسترول بدن
فاکتورهای ارثی (مهمترین نقش) را در میزان سنتز کلسترول بدن به عهده دارند. فاکتورهای دیگر دخیل در میزان سنتز کلسترول شامل رژیم غذایی و فاکتورهای محیطی میباشند. جایگزینی اسیدهای چرب دارای یک (روغنزیتون) یا چند پیوند غیراشباع (روغن ذرت و آفتابگردان) بهجای اسیدهای چرب اشباع (چربی کره، چربی گوشت گاو، روغن خرما) نیز در کاهش سنتز کلسترول، مفید است.
اسیدهای چرب دارای چند پیوند غیراشباع، از طریق افزایش گیرندههای LDL و درنتیجه، افزایش کاتابولیسم آنها، موجب کاهش میزان کلسترول بدن میشوند. اسیدهای چرب اشباع، از طریق تشکیل ذرات کوچکتر VLDL و دارای کلسترول نسبتاً بیشتر، عمل میکنند؛ زیرا این ذرات کوچکتر، نسبت به ذرات بزرگتر VLDL، با سرعت کمتری مصرف شده و درنتیجه، موجب افزایش سطح کلسترول خون میشوند. مصرف سوکروز و فروکتوز نیز نسبت به سایر کربوهیدراتها منجر به افزایش لیپیدهای خون و بهویژه تری آسیل گلیسرولها، خواهد شد.
افزایش کلسترول (هیپر کلسترولمی)
افزایش کلسترول خون در مواردی از قبیل هایپرکلسترولمی فامیلی تیپ 2، کلستاز، نارسایی مزمن کلیه، دیابت شیرین کنترل نشده، الکلیسم، چاقی و … دیده میشود. بارداری معمولاً با افزایش سطح کلسترول همراه است. در یرقانهای انسدادی به علت اختلالاتی که در ترشح و دفع صفرا حاصل میشود، کلسترول به خون بازگشته و مقدار آن افزایش پیدا میکند. در نفروزها، دفع زیاد پروتئین توسط ادرار باعث کاهش یافتن فشار انکوتیک پلاسما شده كه برای جبران این کاهش، مقدار لیپیدهای خون و بهخصوص کلسترول افزایش مییابد و ممکن است تا mg/dl 1000 نيز برسد. در دیابت به علت اختلال در متابولیسم قندها، چربیهای بدن آزاد شده كه تجزيه ناقص آنها باعث پیدایش ترکیبات کتونی و نیز افزایش کلسترول خون میگردد. در هیپوتیروئیدی، درنتیجه سوختوساز كاهشيافته که به دنبال آن افزایش کلی لیپیدها را خواهیم داشت.
کاهش کلسترول (هیپو کلسترولمی)
کاهش کلسترول خون در مواردی از قبیل نقص α- لیپوپروتئین، نئوپلاسم بدخیم کبد، هیپرتیروئیدی، سوءتغذیه، سوختگی شدید و … مشاهده میشود. به دلیل اینکه کبد مواد دارای کلسترول را متابولیزه میکند، سطح غیرطبیعی پایین کلسترول نشاندهندهی بیماریهای شدید کبد است. به دلیل اینکه منبع اصلی کلسترول رژیم غذایی است، سوءتغذیه هم با سطوح پایین کلسترول همراه خواهد بود.
مبتلایان به سکتهی قلبی حاد (AMI) ممکن است به مدت 8-6 هفته تا 50 درصد کاهش کلسترول داشته باشند. در هیپرتیروئیدی با افزایش متابولیسم پايه مقدار لیپیدها و کلسترول خون کم میگردد.
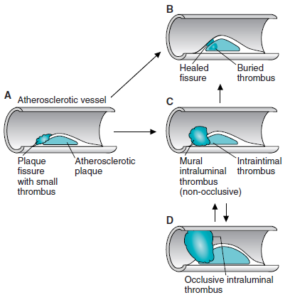
ارتباط کلسترول سرم با بروز آترواسکلروز
افزایش سطح کلسترول پلاسما بهعنوان یک فاکتور اصلی ایجاد آترواسکلروز میباشد. تری آسیل گلیسرولها یک ریسک فاکتور مستقل میباشند. آترواسکلروز شامل رسوب کلسترول و استر کلسترول لیپوپروتئینهای پلاسما در دیواره شریانی است. بروز آترواسکلروز زودرس یا بسیار شدید در بیماریهای با سطوح بالای VLDL، IDL، باقیماندههای شیلومیکرون یا LDL به مدت طولانی (دیابت ملیتوس، هیپوتیروئیدی، نفروز لیپیدی و …) همراه میباشد. یک ارتباط معکوس بین غلظت HDL (HDL2) و بیماری عروق کرونر قلب وجود دارد.
معادله فريدوالد
![]()
در مواردي كه سطح TG بيش از mg/dl 400 ميباشد، نميتوان از معادله فريدوالد براي محاسبه ميزان LDL استفاده نمود و بايد ميزان LDL را بهطور مستقيم اندازهگيري نمود. بهطورکلی، مطالعات نشان ميدهند كه به ازاي هر mg/dl 1 كاهش در LDL پلاسما، مرگومیر ناشي از بيماريهاي آترواسكلروزي قلبي در حدود 2% كاهش مييابد.
فاكتورهاي خطر اصلي براي تعديل هدفهاي LDL
- استعمال دخانيات
- هيپرتانسيون (140/90 BP ≥ با داروهاي آنتيهيپرتانسيون)
- HDL-C پايين (mg/dl 40 >)
- تاريخچه فاميلي CHD
- سن (45 ≤ مردان؛ 55 ≤ زنان)
- عارضه ديابت
- CHD موجود از قبل
سطح هدف LDL در وضعیتهای مختلف
- بیماران دارای بيماري كرونري قلب و يا ديابتيک: كمتر از mg/dl 100
- افراد فاقد هر كدام از ريسك فاكتورها: نگه داشتن در محدوده كمتر از mg/dl 160
- افرادي داراي 2 و يا بيشتر از ريسك فاكتورها: كمتر از mg/dl 130
تعيين نسبت LDL به HDL (LDL/HDL Ratio)
اين نسبت از تقسيم ميزان LDL به HDL به دست ميآيد. ميزان ايده آل اين نسبت در محدوده كمتر از 3 ميباشد.
| Risk Level | LDL/HDL Ratio |
| Low risk Average risk Moderate risk High risk |
3.3 – 4.4 4.4 – 7.1 7.1 – 11.0 11.0 |
تعيين نسبت HDL به LDL (HDL/LDL Ratio)
اين نسبت از تقسيم ميزان HDL به LDL به دست میآید. چنانچه اين نسبت بيشتر از 0.3 باشد، گفته ميشود كه شخص ازنظر پروفايل ليپيدي سالم ميباشد.
| Risk Level | HDL/LDL Ratio |
| Low risk Average risk Moderate risk High risk |
0.22 – 0.30 0.14 – 0.22 0.09 – 0.14 0.09 |
انواع لیپوپروتئینها و ویژگیهای آنها:
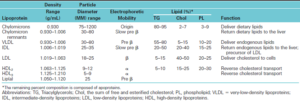
اختلالات اولیه لیپوپروتئینهای پلاسما:
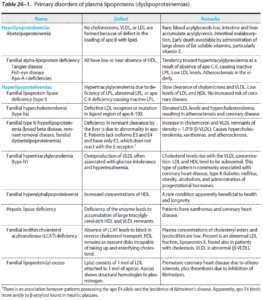
طبقهبندی فردریکسون برای هیپرلیپیدمی ها
به نوع لیپوپروتئین افزایش یافته در هر کدام از دستههای هیپرلیپیدمی در جدول زیر توجه شود.
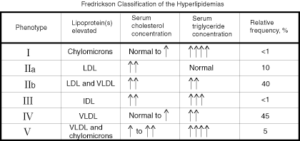
هیپرکلسترولمی پلی ژنیک (غیر فامیلی)
تقریباً 85% هیپرکلسترولمی در جمعیت میتواند در این دسته قرار گیرد. این واژه برای توصیف بیمارانی به کار میرود که افزایش کلسترول وابسته به سن داشته و به تعدیلات در سبک زندگی، پاسخ نمیدهند.
هیپرکلسترولمی فامیلی (FH) (اتوزوم غالب)
یک ناهنجاری اتوزومی غالب با جهش در ژن رسپتور LDL میباشند. FH هتروزیگوت، با شیوع 1 در 500 نفر (آترواسکلروز زودرس) دیده میشود. بروز این بیماری در مردهای هتروزیگوت متأثر شده معمولاً در دهه چهارم زندگی رخ میدهد. در زنان 15- 10 سال دیرتر مشخص میشود. ظهور FH هموزیگوت در کودکی (سطح LDL بیشتر از 400) رخ میدهد.
نقص فامیلی آپو B
یک ناهنجاری اتوزومی غالب با شیوع 1 در 750 میباشد. این نقص موجب ممانعت از تشخیص آپو B100 بهوسیله رسپتور LDL میگردد. معایب فیزیکی در افراد مبتلا، مشابه علائم افراد در بیماری FH میباشد.
سیتواسترولمی
یک ناهنجاری نادر اتوزومی بوده که با جذب و تجمع فیتواسترول ها (استرولهای گیاهی) در پلاسما و بافتهای کناری همراه میباشد. در این اختلال، جهش در ژنهای ABCG8 و ABCG5 وجود دارد. در این ناهنجاری، اختلال در پمپ کردن غیرفعال فیتواسترول ها به درون روده و به درون صفرا وجود دارد.
دیسلیپیدمی دیابتی
این اختلال، متشکل از دیسلیپیدمی آتروژنیک (TG بالا، HDL پایین و LDL بالا) در افراد دارای دیابت نوع دو میباشد. در این مورد، درمان اغلب به کاهش سطح LDL معطوف میگردد.
هیپرتری گلیسریدمی فامیلی
ناهنجاری معمولاً در بلوغ با سطح TG ناشتا در محدوده 500- 200 میلیگرم در دسیلیتر رخ میدهد. در این بیماری، افزایش تولید VLDL در حضور تولید طبیعی آپو B وجود دارد.
نقص LPL
یک ناهنجاری نادر اتوزومی مغلوب میباشد. تظاهر در کودکی و با درد شکمی و پانکراتیت همراه است. این نقص موجب ایجاد سندروم شیلومیکرونمی کلاسیک نوع یک میگردد. سطوح ناشتای TG میتواند بیشتر از 100 و پس از مصرف غذا بیشتر از 10000 میلیگرم در دسی لیتر باشد.
نقص آپو C-II
آپو CII، فاکتور فعال کننده LPL میباشد. یک ناهنجاری نادر اتوزومی مغلوب بوده و با بروز ناهنجاری در کودکان و بالغین جوان با دورههای عودکننده از درد شکمی و پانکراتیت همراه میباشد.
افزایش آپو C-III
آپو CIII، باعث تداخل با فعالیت LPL میگردد. ممانعت آن از طریق اتصال به انتهای کربوکسی آپولیپوپروتئین B و ممانعت از اتصال به رسپتور LDL رخ میدهد.
هیپرلیپیدمی ترکیبی فامیلی (نوع 2B)
شیوع این اختلال، 1 در 100 نفر میباشد. فامیلهای متأثر شده باید بیش از یک الگوی ناهنجاری لیپیدی داشته باشند. افراد متأثر شده میتوانند هیپرکلسترولمی ساده، هیپرتری گلیسریدمی ساده و یا نقص مختلط داشته باشند.
دیس بتا لیپوپروتئینمی (نوع III)
شیوع این اختلال، 1 در 100 نفر میباشد. در این بیماری، نقص در آپو E وجود دارد. آپو E در ساختمان شیلومیکرون ها، باقیمانده شیلومیکرون ها، VLDL و IDL وجود دارد و به رسپتور LDL متصل میشود. اساساً افراد بالغ را متأثر میسازد. در این بیماری، افزایش کلسترول و TG، تقریباً بهطور یکسان خواهیم داشت. در الکتروفورز این بیماران، وجود باند پهن غیرطبیعی بین VLDL و LDL خواهیم داشت.
نقص لیپاز کبدی (HL)
یک ناهنجاری نادر فامیلی با جهش در ژن HL بوده که همراه با هیپرلیپیدمی مرکب میباشد. سطوح کلسترول بین 1500- 250 و TG بین 8000- 400 میلیگرم در دسی لیتر میباشد.
آبتا لیپوپروتئینمی
یک ناهنجاری نادر اتوزومی مغلوب میباشد. در این بیماری، تخریب آپو B، مدت کوتاهی پس از ترجمه (نقص در پروتئینهای ناقل میکروزومی) رخ میدهد. فقدان آپو B48 و B100 در پلاسمای این بیماران وجود دارد.
هیپوبتا لیپوپروتئینمی
یک ناهنجاری اتوزومی غالب میباشد. این بیماری، در برخی از فامیلها با جهشهای Nonsense یا Missense در ژن آپو B توجیه میشود. در این افراد، جذب بد چربی و سطوح پایین کلسترول خواهیم داشت. میزان کلسترول کمتر از 50 میلیگرم در دسیلیتر در افراد هموزیگوت مشاهده میشود. این افراد با کاهش خطر بیماری قلبی- عروقی همراه میباشند.
بیماری ابقاء شیلومیکرون (Chylomicron retention disease)
در این بیماری، تنها آپو B48 متأثر شده و در کودکی و با سوء جذب چربی ظاهر میگردد.
هیپوآلفا لیپوپروتئینمی
یک ناهنجاری اتوزومی غالب با شیوع 1 در 400 میباشد. سطح HDL این بیماران کمتر از 30 در مردان و کمتر از 40 در زنان متأثر شده میباشد. در این بیماران، نقص در لیپاز کبدی، نقص در ژن آپو A-IV، نقص در ژن آپو A-I، نقص در ژن آپو C-III و جهش در ژن ABCA1 وجود دارد.
نقص آپو A-I و آپو C
یک وضعیت اتوزومی مغلوب نادر با کاهش در تشکیل HDL میباشد. اغلب سطح HDL کمتر از 5 میلیگرم در دسی لیتر میباشد.
بیماری تانژیر
یک ناهنجاری نادر اتوزومی مغلوب میباشد. در این افراد، کلسترول پایین و TG بالا، HDL کم و یا غیرقابلشناسایی خواهیم داشت. بیماران دارای لوزههای نارنجی بوده و جهش در ژن ABCA1 وجود دارد.
نقص LCAT
این بیماری با جهش در ژن LCAT همراه میباشد و به 2 فرم رخ میدهد:
1- نقص LCAT فامیلی کلاسیک (کامل)
2- نقص معتدلتر جزئی معروف به بیماری چشم ماهی (Fish eye disease)
بیماری چشم ماهی (Fish eye disease)
نقص ژن پروتئین ناقل استر کلستریل (CETP)
یک ناهنجاری اتوزومی مغلوب بوده و همراه با مهار انتقال استرهای کلسترول میباشد. CETP در انتقال استر کلستریل از HDL به لیپوپروتئینهای غنی از آپو B100 (VLDL و LDL) در معاوضه با TGها نقش دارد. وجود ذرات HDL بزرگ و مملو از استر کلسترول و افزایش HDL به بیش از 100 میلیگرم در دسیلیتر در این بیماران خواهیم داشت.
آزمایش پلاسمای ساکن (Standing plasma test)
مقداری از پلاسما در یک لوله آزمایش ریخته و در یخچال 4 درجه سانتیگراد به مدت یکشب قرار میدهیم. شیلومیکرونها بهصورت یکلایه خامه شناور در سطح نمونه انباشته میگردند. وجود VLDL ایجاد کدورت مینماید. در صورت ایجاد لایه خامه مانند و کدورت (هر دو)، وجود شیلومیکرون ها و VLDL (هر دو) در نمونه تائید میگردد.
تشخیص VLDL و LP(a)
هر دو لیپوپروتئین در الکتروفورز، در ناحیه بتا حرکت میکنند. برای تشخیص VLDL، الکتروفورز بخش اولتراسانتریفوژی با دانسیته کمتر از 1.006 kg/L و برای تشخیص LP(a)، الکتروفورز بخش اولتراسانتریفوژی با دانسیته بیشتر از 1.006 kg/L باید صورت گیرد. اگر مقدار LP(a) بیش از 30- 20 میلیگرم در دسی لیتر باشد، یک باند اضافی با تحرک پره بتا نیز در بخش اولتراسانتریفوژی با دانسیته بیشتر از 1.006 kg/L مشاهده میشود که Sinking pre-beta lipoprotein نام دارد. در این شرایط، ممکن است پزشک درخواست اندازهگیری کمی LP(a) نماید.
تغييرات بيولوژيك
ميانگين ضريب تغييرات فيزيولوژيك براي كلسترول در يك فرد در حدود 6/5 درصد و تغییر سطوح كلسترول در 95 درصد نمونهها تا حدود 13 درصد بالا يا پایینتر از سطح ميانگين فرد میباشد. سطوح كلسترول در زمستانها كمي بيشتر است. اثر تغييرات غذايي چندين هفته طول ميكشد تا ظاهر گردد. بنابراين قبل از مطمئن شدن از سطح كلسترول فرد اهميت دارد كه براي دو هفته رژيم غذايي معمولي داشته و نه اضافهوزن و نه كاهش وزن داشته باشد.
داروهايي از قبيل ضدبارداریهای خوراكي، استروژنهاي پس از يائسگي و برخي از داروهاي ضد افزايش فشارخون موجب تغيير در سطوح ليپيدي ميگردند. شيوه زندگي و فاكتورهاي بيولوژيك كه تغييرات کوتاهمدت از مقادير پايه ليپيدي را ايجاد مينمايند شامل ناشتايي، وضعيت بيمار، انسداد رگ، ضد انعقادها، سكته قلبي اخير، سكته مغزي، كاتتريزاسيون قلبي، تروما، عفونت حاد و بارداري ميباشند. توصيه شده است كه اندازهگیری ليپوپروتئينها زودتر از 8 هفته بعد از هرگونه تروما يا عفونت حاد باكتريايي و ويروسي و 4-3 ماه پس از زايمان صورت نگيرد.
ناشتايي
بهطور ايده آل فرد بايد به مدت 12 ساعت قبل از نمونهگیری، ناشتا باشد. شيلوميكرونها معمولاً در پلاسماي پس از غذا، بسته به نوع و ميزان غذاي بلع شده وجود داشته و ميتوانند بهطور قابلملاحظهاي غلظت تريگليسريد پلاسما را افزايش دهند. شيلوميكرونها طي 9-6 ساعت تقريباً کاملاً پاکشده و حضورشان پس از 12 ساعت ناشتايي غیرطبیعی به نظر ميرسد. بهطورکلی، سطوح TC و HDL را ميتوان در افراد غير ناشتا اندازهگيري نمود كه اين كار سبب تسهيل غربال كردن و مانيتور كردن ميگردد.
ناشتايي اثر کمی روی سطح TC پلاسمايي دارد و اگرچه سطوح غير ناشتاي HDL ميتواند چند mg/dl كمتر از سطوح ناشتا باشد، اما اين وضع نبايستي به طبقهبندي اشتباه بيمار با سطوح پايين HDL منجر گردد. وقتیکه TG و LDL-C اندازهگيري ميشوند، ناشتايي لازم ميشود. وجود شيلوميكرونها پس از تغذيه و تغييرات در LDL منجر به تخمين كمتر از حد LDL-C شده و ميتواند منجر به طبقهبندي اشتباه بيماران گردد.
بهطورکلی ميتوان گفت كه براي انجام آزمايشهاي كلسترول وHDL ميتوان از نمونههاي غيرناشتا هم استفاده نمود اما براي اندازهگيري ميزان تريگليسريد و LDL بايد حتماً فرد ناشتا باشد. همچنين ميتوان براي ارزيابي وضعيت پروفايل ليپيدي در بيمار و در صورت ضرورت و اضطرار از نمونههاي غيرناشتا استفاده نموده و فقط آزمايشهاي كلسترول و HDL را درخواست نمود. در اين موارد چنانچه ميزان كلسترول بيمار مساوي و يا بيشتر از 200 و ميزان HDL نيز كمتر از mg/dl 40 بود، لازم است كه در وضعيت ناشتا نيز مجدداً پروفايل ليپيدي شخص بررسي گردد و در غير اين صورت نيازي به اين كار نميباشد.
وضعيت بدن در هنگام نمونهگيري
هنگامیکه وضعيت بيمار از حالت ايستاده به حالت درازكش تغيير مييابد، آب خارج رگي به سيستم عروقي منتقلشده و اجزاي پلاسمايي غیرقابلانتشار را رقيق مينمايد. كاهشي به بزرگي 10% در غلظت TC، LDL، HDL، آپو A-I و آپو B پس از 20 دقيقه دراز كشيدن مشاهده شده است. كاهش در TG حدود 50% بيشتر بوده كه پيشنهاد ميكند فاكتورهايي غير از رقيق شدن خون نيز ميتوانند در اين امر دخيل باشند. اين تغييرات در فردي كه از حالت ايستاده به حالت نشستن تغيير وضعيت ميدهد، نصف ميگردد. دستورالعملهاي اخير NCEP توصيه مينمايد كه بيمار به مدت 5 دقيقه قبل از نمونهگيري بهمنظور ممانعت از تغليظ خوني بنشيند. انسداد طولاني رگ ميتواند منجر به تغليظ خون و افزايش 15-10 درصدي در كلسترول گردد.
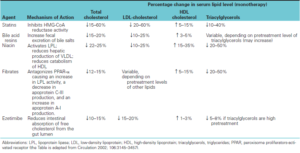
داروهای کاهشدهنده لیپیدهای خون
داروهای کاهشدهنده لیپیدهای خون، از طریق مکانیسمهای مختلفی، موجب کاهش سطح لیپیدها شده که در جدول زیر آمدهاند.
منابع:
1-Tietz Textbook of Clinical Chemistry and Molecular Diadnosis. 2006; 4th Edition.
2- Henrys Clinical Diagnosis and Management by Laboratory Methods. 2007; 21st Edition.
3 – Wendy Aineson and Jean Brickell. Clinical Chemistry; A Laboratory Perspective. 2007; 1st Edition.
4- Arneson W, Brickell J. Clinical chemistry; a laboratory perspective. 2007.
5- Pagana KD and Pagana TJ. Diagnostic and laboratory test refrence. 2005; 7th Edition.
6- Van Leeuwen AM, Kranpitz TR and Smith L. Laboratory and diagnostic tests with nursing implications. 2006 ; 2nd Edition.
7- Wilson DD. Manual of laboratory and diagnostic tests. 2008.
اهميت و اندازهگيري ليپيدهاي سرم
https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/cholesterol-metabolism
https://medlabnews.ir/%da%a9%d9%84%d8%b3%d8%aa%d8%b1%d9%88%d9%84-%d8%a8%d8%af/
https://medlabnews.ir/%d9%84%db%8c%d9%be%db%8c%d8%af/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام