متابولیسم قندها و بیماریهای ناشی از اختلالات آنها
مراد رستمي: کارشناس ارشد بیوشیمی بالینی، دانشگاه علوم پزشكي جندیشاپور اهواز
معصومه جرفی: کارشناس ارشد میکروبشناسی، دانشگاه علوم پزشكي جندیشاپور اهواز
مسیر پنتوز فسفات
سرنوشت اصلی گلوکز در پستانداران، ورود به سیکل گلیکولیز و تولید پیروات و ورود به سیکل کربس و در نهایت تولید ATP میباشد. مسیر پنتوز فسفات (که فسفوگلوکونات و یا هگزوز منوفسفات نیز نامیده میشود)، نیز یکی از راههای متابولیسمی گلوکز بوده که به دلیل تولید NADPH (در مرحله اکسیداتیو بازگشتناپذیر) و ریبوز (در مرحله غیراکسیداتیو بازگشتپذیر)، دارای اهمیت میباشد. از NADPH بهعنوان یک احیاءکننده در مسیرهای آنابولیک (مانند بافتهای فعال در سنتز اسیدهای چرب و استروئیدها مانند غدد پستان، قسمت قشری غده فوق کلیوی، کبد و بافت چربی) و همچنین در احیاء گلوتاتیون اکسیده به فرم احیاء استفاده میشود. از ریبوز نیز در سنتز اسیدهای نوکلئیک استفاده میشود. بافتهای دارای فعالیت کمتر در سنتز اسیدهای چرب (نظیر عضله اسکلتی)، فاقد مسیر پنتوز فسفات هستند.

کمبود گلوکز 6- فسفات دهیدروژناز (Glucose 6-phosphate dehydrogenase)
کمبود آنزیم گلوکز-6- فسفات دهیدروژناز (G6PD) شایعترین بیماری ناشی از کمبود آنزیم در دنیا بوده و تقریباً 400 میلیون نفر در سراسر جهان به این بیماری مبتلا هستند. موتاسیونهای ژن کدکننده G6PD میتوانند موجب تولید آنزیمی با فعالیت کمتر و یا پایداری کمتر شوند. تاکنون بیش از 440 واریانس مختلف در ژن G6PD شناسایی شده که با درجات مختلفی از نقص در عملکرد این آنزیم همراه هستند.
انرژی موردنیاز گلبولهای قرمز که در فعالیت غشاء سلول و محافظت از اجزای داخلی آن نقش دارند از طریق متابولیسم گلوکز تأمین میشود که 90 درصد آن بهصورت متابولیسم بیهوازی و از مسیر امبدن میرهوف (گلیکولیز) به انجام میرسد. 10 درصد باقیمانده نیز بهصورت هوازی و با استفاده از مسیر پنتوز فسفات حاصل میشود. G6PD یک آنزیم کلیدی در چرخه متابولیسم گلوکز بوده که در طی تبدیل گلوکز 6ـ فسفات به 6ـ فسفو گلوکونولاکتون، موجب تولید NADPH نیز میشود. در حالت عادی، در هنگام مواجهه گلبولهای قرمز با داروها و سموم تولیدکننده رادیکالهای اکسیژن، فعالیت آنزیم G6PD نیز تا چندین برابر افزایش یافته و بدین طریق از تخریب هموگلوبین و غشای گلبولهای قرمز توسط این مواد، جلوگیری مینماید.
اين آنزيم بهعنوان يك آنزيم محافظ براي گلبولهای قرمز و طول عمر طبيعي آنها ضروري بوده و كاهش و یا فقدان آن، سبب تخريب گلبولهای قرمز و در نتيجه، ایجاد یک نوع از کمخونی موسوم به آنمي هموليتيك میشود. از آن جایی که آلل ژن این آنزیم روی کرموزوم X قرار دارد، بنابراین کمبود G6PD یک اختلال وابسته به جنس محسوب میشود.
تخريب گلبولهای قرمز معمولاً در اثر خوردن باقلا و برخی از داروهای اکسیدان و یا ابتلا به برخی از عفونتهای باکتریایی و یا ویروسی، آغاز ميشود. در برخی از گزارشهای موردی، نشان داده شده است که قرار گرفتن برخی از بیماران فاویسمی در معرض بوی حنا نیز باعث ایجاد همولیز در آنها شده است. همچنین نشان داده شده است که برخی از بیماران دچار کمبود و یا فقدان G6PD، پس از مصرف باقلا و یا داروهای اکسیدان، دچار همولیز نمیشوند. بررسیها نشان داده است که این افراد دارای مقادیر بالایی از آنزیم کاتالاز بوده و سطوح بالای این آنزیم، کمبود و یا فقدان آنزیم G6PD را جبران مینماید.
در طی یک بررسی، بین سطوح بالاتر بیلیروبین در نوزادان متولد شده، با کمبود G6PD، یک ارتباط مستقیم مشاهده شده است.
| گروه نوزادان تازه متولد شده | تعداد (نفر) | درصد کمبود G6PD |
| طبیعی | 500 | 22/5 |
| زردی ملایم (میزان بیلیروبین 20- 15 میلیگرم در دسیلیتر) | 38 | 45 |
| زردی ملایم (میزان بیلیروبین بیشتر از 23 میلیگرم در دسیلیتر) | 70 | 60 |
| بستریشده با علامت کرنیکتروس | 20 | 78 |
مکانیسم عمل آنزیم G6PD:
در شرایط طبیعی، مواد اکسیدان بهسرعت توسط گلوتاتیون احیاء (GSH) و با فعالیت گلوتاتیون پراکسیداز، غیرفعال میشوند که منجر به تبدیل گلوتاتیون احیاء (GSH) به گلوتاتیون اکسیده (GSSG) میشوند. گلوتاتیون اکسیده (GSSG) از طریق گلوتاتیون ردوکتاز و با کمک NADPH مجدداً به گلوتاتیون احیاء (GSH) تبدیل میشود. گلوتاتیون پراکسیداز به سلنیوم بهعنوان کوفاکتور احتیاج دارد. ارتباط بین برخی سرطانها با میزان پایین سلنیوم و کاهش فعالیت گلوتاتیون پراکسیداز در خون گزارش شده است.

در کمبود G6PD گلبولهای قرمز نمیتوانند در هنگام مواجهه با مواد اکسیدان، NADPH کافی تولید نموده و موجب بازیابی گلوتاتیون احیاء (GSH) از گلوتاتیون اکسیده (GSSG) شوند؛ لذا قابلیت سلول در حذف H2O2 یا رادیکالهای آزاد از بین رفته و هموگلوبین، اکسیده شده و بهصورت اجسام هاینز (Heinz body) در داخل گلبولهای قرمز رسوب مینماید؛ در نتیجه، گلبولهای قرمز در طحال تخریب شده و موجب ایجاد کمخونی میگردند.
انواع G6PD:
در زیر به اختصار به برخی از انواع مهمتر G6PD اشاره میگردد:
1) G6PD-B : نوع طبیعی آنزیم G6PD به نام G6PD-B نامیده میشود.
2) +G6PD-A: در 20% از سیاهپوستان دیده میشود. این نوع از آنزیم دارای عملکرد طبیعی میباشد.
3) –G6PD-A دسته دیگری هستند که فقط 15% آنزیم طبیعی G6PD، فعالیت داشته و در ۱1% از سیاهپوستان آمریکایی دیده میشود. در این نوع از آنزیم، فعالیت آنزیمی G6PD در گلبولهای قرمزی که عمرشان به ۴۰ روز میرسد، بهسرعت کاهش مییابد، درصورتیکه فعالیت G6PD طبیعی پس از 120 روز عمر گلبول قرمز، به میزان 50% خود، کاهش مییابد.
4) G6PD-Med که در ناحیه مدیترانه مشاهده شده و شدیدتر از سایر انواع میباشد.
5) G6PD-Canon که یک ژنوتایپ غیرطبیعی بوده و در تایلند، ویتنام و سایر جمعیتهای آسیایی مشاهده میشود.
6) سایر انواع G6PD: در جمعیت مدیترانهای و چینی، شایعتر هستند.
بیشترین شیوع این بیماری، در آفریقا و سپس در آسیا و بهویژه در ناحیه مدیترانه دیده میشود.
انواع G6PD بر اساس طبقهبندی WHO:
سازمان جهانی بهداشت (WHO)، به طبقهبندی انواع G6PDهای مختلف بر اساس میزان کمبود آنزیم و شدت همولیز پرداخته است.
کلاس I: نادر بوده و کاهش آنزیم در آنها شدید (کمتر از 10% فعالیت طبیعی آنزیم) میباشد. این افراد دارای آنمی همولیتیک مزمن هستند.
کلاس II: در این دسته نیز کاهش آنزیم شدید بوده، اما اغلب دارای همولیز متوسطی (intermittent hemolysis) میباشند.
کلاس III: دارای کاهش آنزیم با شدت متوسط (10 تا 60% فعالیت طبیعی آنزیم) بوده و در صورت مواجهه با عفونت و یا مصرف داروهای اکسیدان، همولیز متوسطی (intermittent hemolysis) پیدا میکنند.
کلاس IV: فاقد کاهش آنزیم و یا همولیز هستند.
کلاس V: در این دسته، فعالیت آنزیمی افزایش یافته است.
کلاسهای IV و V: از نظر کلینیکی فاقد اهمیت هستند.
شدت بیماری G6PD به میزان کاهش این آنزیم بستگی دارد:
| میزان فعالیت آنزیم G6PD | شدت بیماری |
| کمتر از ۱۰% میزان طبیعی | بیماری شدید |
| بین ۱۰% تا ۶۰% میزان طبیعی | بیماری با شدت متوسط |
| بیشتر از ۶۰% میزان طبیعی | فاقد علامت |
۱۰ تا ۱۵% سیاهپوستان آمریکایی به این بیماری مبتلا هستند. فرد بیمار (که دچار کمبودG6PD است) در هنگام مواجهه با مواد اکسیدان (بعضی از داروها، عفونت و باقلای مازندرانی) دچار تخریب هموگلوبین و در نتیجه رسوب آن در گلبول قرمز شده و اجسامی به نام اجسام هاینز (Heinz Bodies) ایجاد میشود. مواد اکسیدان موجب آسیب به غشای گلبولهای قرمز و ایجاد همولیز داخل عروقی (که بهصورت هموگلوبین در ادرار دیده میشود) و همولیز خارج عروقی (که بهصورت بزرگی طحال و زردی نمایان میشود) میشوند.
علاوه بر برخی از خوراکیها مانند باقلا، برخی از بیماریها مانند اسیدوز دیابتی، نارسایی حاد کلیوی و هپاتیت نیز میتوانند منجر به بروز همولیز در افراد مستعد شوند. داروها و ترکیباتی از قبیل سولفامتوکسازول، سولفانیلامید، سولفاپیریدین، کلروکوئین، پاماکوئین، پنتاکوئین، نیتروفورانتوئین، نالیدیکسیک اسید، فنازوپیریدین، سیپروفلوکساسین، نورفلوکساسین، کلرامفنیکل، نفتالن، آنالوگهای ویتامین K، متیلنبلو، استانیلید، دوکسوروبیسین، ایزوبوتیل نیتریت و فنیل هیدرازین نیز میتوانند با خطر بروز همولیز در افراد دچار کمبود G6PD همراه باشند که این نوع از بیماران باید از مصرف اینگونه داروها خودداری نمایند.
علائم بالینی:
بیماران مبتلا به کمبود G6PD در هنگام مواجهه با عوامل اکسیدان، دچار همولیز گلبولهای قرمز میشوند. علائم بیماری شامل کسالت، بیحالی، درد شکم و یا درد کمر، تهوع و استفراغ بوده و پس از چند ساعت تا چند روز (اغلب ۲ تا ۳ روز) (البته شروع علائم در کودکان ناگهانی است)، بیمار دچار زردی و ادرار تیره رنگ میشود (نشانه وجود هموگلوبین در ادرار).
مهمترین خطر این بیماری، نارسایی حاد کلیه (ARF) خصوصاً در کودکان است. کمخونی معمولاً از نوع نرموسیتیک- نرموکرومیک بوده و هموگلوبین بیماران گاهی بهg/dl ۴ و یا حتی کمتر نیز میرسد. کاهش شدید هموگلوبین و هماتوکریت خون، هموگلوبینمی، هموگلوبینوری، کاهش یا فقدان هاپتوگلوبین پلاسما و افزایش درصد رتیکولوسیتهای خون محیطی در آزمایش این افراد مشاهده میشود. لازم به ذکر است که در این افراد، انجام آزمایش G6PD پس از وقوع حملات همولیزی، به دلیل وجود فعالیت بالای این آنزیم در رتیکولوسیتها، میتواند بهصورت کاذب، طبیعی جلوه نماید.
بیماری فاویسم (Favism):
در اغلب مطالعات انجام شده، میزان ناکافی آنزیم G6PD در نوزادان پسر بین 4-3% و در نوزادان دختر، کمتر از 1% گزارش شده است. 8% نوزادان گیلانی مبتلا به کمبود G6PD هستند. مهمترین و شدیدترین شکل آنمی همولتیک، ناشی از کمبود G6PD است. این بیماری در هر سنی میتواند اتفاق بیفتد ولی در کودکان بیشتر دیده میشود. شروع بیماری در بچهها معمولاً همراه با بیقراری، سپس تب، درد شکمی، اسهال و گاهی اوقات استفراغ، بیحالی و سستی (lethargic) میباشد. هموگلوبینوری ظرف ۶ تا ۲۴ ساعت پس از علائم فوق شروع میشود. در معاینه فیزیکی، رنگپریدگی، تاکیکاردی، زردی و بزرگی کبد قابل مشاهده میباشد. در موارد شدید بیماری، شوک هیپوولمیک و در موارد خیلی نادری نیز نارسایی قلبی مشاهده شده است.
علت ایجاد فاویسم به خاطر وجود دو ماده ویسین (Vicine) و کانویسین (Convicine) در باقلا بوده که در بدن، تحت روند اتواکسیداسیون، تولید رادیکالهای آزاد اکسیژن مینمایند. باید توجه داشت که همه اشکال تهیه شده باقلا (پخته، خشک و بوی آن) ایجاد همولیز میکنند. بالغین مبتلا به دلایل نامعلوم دچار حملات حاد فاویسم نخواهند شد (البته به مقدار و کیفیت باقلای خورده شده نیز بستگی دارد). البته همانطوری که قبلاً نیز بیان شد، این افراد احتمالاً دارای مقادیر بالایی از آنزیم کاتالاز بوده و سطوح بالای این آنزیم، کمبود و یا فقدان آنزیم G6PD را جبران مینماید.
علل افزایش G6PD:
کمخونی پرنیسیوز، کمخونی مگالوبلاستیک، خونریزی مزمن، انفارکتوس میوکارد، اغمای کبدی و هیپرتیروئیدی میتوانند سبب افزایش G6PD شوند، بنابراین در این موارد، میزان آنزیم افزایش داشته و کمبود آن اغلب مشخص نمیشود.
علل کاهش G6PD:
کمبود یا نقص آنزیم G6PD و کمخونی همولیتیک غیر ایمونولوژیک نوزادان، سبب کاهش G6PD میشوند.
نمونه های موردنیاز:
نمونه CBC غیرهمولیز. برای انجام این آزمایش، نیازی به ناشتا بودن فرد نیست. برای نمونهگیری در این آزمایش از ضدانعقادهای EDTA و یا هپارین استفاده میشود. استفاده از ضدانعقادهای اگزالات و یا فلوئور در این آزمایش توصیه نمیشود. در مورد این آزمایش از فریز کردن نمونه باید اجتناب شود. آزمایش G6PD در دمای 4 درجه سانتیگراد به مدت 20 روز پایدار است. به دلیل اینکه گلبولهای سفید دارای فعالیت آنزیمی بیشتری نسبت به گلبولهای قرمز هستند، توصیه میشود که آزمایش بر روی گلبولهای قرمز شسته شده انجام شود
روشهای اندازه گیری و اصول آزمایش:
روش تست فلورسانت لکهای (Fluorescent Spot Test) یا روش باتلر (Butler):
گلبولهای قرمز در حالت طبیعی، حاوی مقادیر طبیعی از آنزیم بوده که اگر این سلول را با گلوکز 6ـ فسفات و +NADP مجاور نماییم، تبدیل +NADP بهNADPH انجام و باعث ایجاد خاصیت فلورسانس میشود؛ زیرا NADPH دارای خاصیت فلورسانسی میباشد. میزان فلورسانس متناسب با میزان آنزیم است. این روش، یک روش کیفی مناسب و رایج برای غربالگری آنزیم G6PD بوده که بر اساس آن فعالیت آنزیم بهصورت کافی (Sufficient)، ناکافی نسبی (Partial defficient) و یا ناکافی (Defficient) بیان میشود. معمولاً فعالیت کمتر از 30% آنزیم بهصورت ناکافی گزارش میشود.
روش اسکوربات سیانید:
در این روش خون با محلول سیانید سدیم و اسکوربات سدیم انکوبه میشود. در این مدت از طریق اکسیداسیون اسکوربات، H2O2 تولید شده و از طرفی، سیانید موجب مهار آنزیم کاتالاز میشود؛ بنابراین H2O2 برای اکسیداسیون هموگلوبین در دسترس قرار گرفته و موجب ایجاد متهموگلوبین و در نتیجه ایجاد رنگ قهوهای میشود. این تغییر رنگ در نقص آنزیمی سریعتر مشاهده میشود.
ارزيابي کمي:
بررسي مقدار کمي G6PD از طريق اسپکتروفتومتري انجامپذیر است. اساس اين تست، اندازهگیری مقدار جذب در طولموج 340 نانومتر است که نمايانگر شکلگیری NADPH است. اين تست از طريق مخلوط کردن هموليزات که حاوي گلوکز-6- فسفات میباشد با کوفاکتور +NADP در دمای 30 درجه سانتیگراد و بررسي توليد NADPH انجام میشود. در نهايت، فعاليت آنزيم G6PD بهصورت IU/RBCs و يا IU/Hb گزارش میگردد. در بررسیهای کمی، مقدار طبیعی این آنزیم معمولاً U/gr hemoglobin 8/8- 13/4 میباشد.
تشخيص مولکولي:
تشخیص مولکولي میتواند براي غربالگري جمعيتي، بررسیهای خانوادگي و تشخيص پيش از تولد مورد استفاده قرار گيرد. بيشترين اهميت تستهای مولکولي جهت آناليز G6PD در زنان هتروزيگوت میباشد
.مداخله کننده ها:
رتیکولوسیتوز قابلتوجه میتواند نتایج مثبت کاذب در این آزمایش ایجاد کند. در مواردی که فرد دچار همولیز شده باشد (مانند افراد فاویسمی که باقلا و یا داروهای اکسیدان مصرف کرده باشند)، توصیه میشود که در طی مراحل مختلف آزمایش و قبل از انتقال نمونه بر روی کاغذ صافی، برداشتن نمونه از ته لوله صورت گیرد؛ زیرا در این بیماران دچار همولیز، تعداد رتیکولوسیتها افزایش یافته و از آن جایی که فعالیت آنزیم G6PD در رتیکولوسیتها، بالاتر است، برداشتن نمونه از لایههای رویی میتواند منجر به نتیجه مثبت کاذب (طبیعی نشان دادن فعالیت آنزیم G6PD) در این بیماران شود؛ زیرا رتیکولوسیتها نسبت به گلبولهای قرمز، دارای وزن مخصوص کمتری بوده و در سطح نمونه قرار میگیرند. خشک نشدن لکه خون روی کاغذ صافی نیز از جمله علل ایجاد نتایج منفی کاذب در این آزمایش به شمار میرود.
آنزیمهای ترانسکتولاز و ترانسآلدولاز
ترانسکتولاز که یک آنزیم وابسته به TPP (تیامین پیروفسفات) و +Mg2 است، در انتقال یک قطعه 2 کربنه (کربنهای 1 و 2) گزیلولوز 5- فسفات به ریبوز 5- فسفات و تولید محصول 7 کربنه سدوهپتولوز 7- فسفات و گلیسرآلدئید 3- فسفات نقش دارد.
ترانسآلدولاز با برداشت یک قطعه 3 کربنه (کربنهای 1 تا 3) از سدوهپتولوز و ترکیب آن با گلیسرآلدئید 3- فسفات، تولید فروکتوز 6- فسفات و اریتروز 4- فسفات مینماید.
حال با عمل مجدد ترانسکتولاز از اریتروز 4- فسفات و گزیلولوز 5- فسفات، محصولهای فروکتوز 6- فسفات و گلیسرآلدئید 3- فسفات تولید میشود.
از اندازهگیری فعالیت ترانسکتولاز به منظور برآورد میزان کمبود تیامین استفاده میشود.

اختلالات ناشی از نقص در مسیر اسید اورونیک:
تولید اسید گلوکورونیک
در مسیر اسید اورونیک، اسید گلوکورونیک تولید میشود که برای کنژوگه شدن با بیلیروبین و گزنوبیوتیکها فرم فعال آن یعنی UDP- گلوکورونات موردنیاز است. برای تسریع ورود گلوکز به مسیر اسید اورونیک میتوان از داروهای باربیتال یا کلروبوتانول در موش صحرایی کمک گرفت.
عدم تولید اسید آسکوربیک در پریماتها
در مسیر اسید اورونیک، L- گولونات تولید شده که پیشساز مستقیم اسید آسکوربیک میباشد. پریماتها (از جمله انسان)، خوکچه هندی و …، به دلیل فقدان وجود L- گولونولاکتون اکسیداز، قادر به ساخت اسید آسکوربیک نمیباشند.
پنتوزوری اولیه
در صورت نقص در آنزیم L-گزیلولوز ردوکتاز، L-گزیلولوز به گزیلیتول تبدیل نشده و باعث ایجاد بیماری پنتوزوری اولیه میشود که با ظاهر شدن مقادیر قابلتوجهی از L-گزیلولوز در ادرار همراه میباشد. به منظور افزایش دفع L-گزیلولوز در بیماران پنتوزوریک میتوان از آمینوپیرین و آنتیپیرین استفاده نمود.

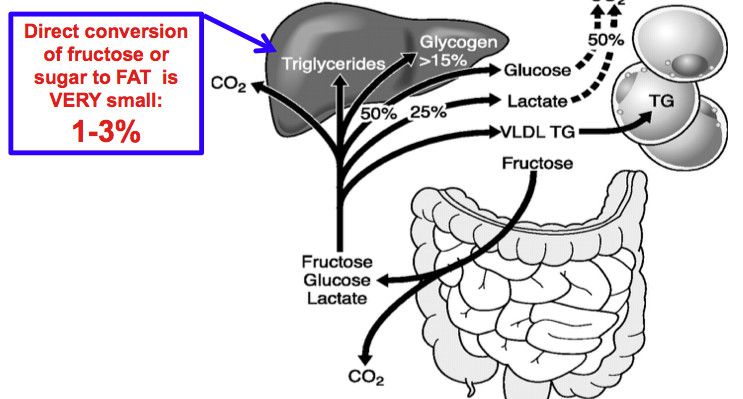
اختلالات ناشی از نقص در متابولیسم فروکتوز:
فروکتوز از متابولیزه شدن سریعتری در کبد نسبت به گلوکز برخوردار میباشد. فروکتوز موجب تقویت ساخت اسیدهای چرب، تریآسیل گلیسرولها، ترشح VLDL، افزایش غلظت LDL (بالقوه آتروژن بودن فروکتوز) و هیپراوریسمی میشود.
فروکتوزوری اولیه (فروکتوزوری اساسی):
یک اختلال اتوزومی مغلوب بوده که در نتیجه کمبود فروکتوکیناز به وجود میآید. فروکتوکیناز مسئول تبدیل فروکتوز به فروکتوز 1- فسفات میباشد. این بیماری معمولاً بدون علامت بوده و ممکن است بهطور اتفاقی بهوسیله شناسایی فروکتوز در ادرار مشاهده شود. درمان خاصی لازم ندارد.
عدم تحمل ارثی فروکتوز:
این اختلال در اثر کمبود فعالیت آلدولاز B که مسئول تبدیل گلیسرآلدئید 3- فسفات به فروکتوز 1 و 6- بیسفسفات میباشد، رخ میدهد. مصرف فروکتوز موجب تجمع فروکتوز 1- فسفات شده که باعث ایجاد علائمی از قبیل هیپوگلیسمی، تهوع و استفراغ میشود. این علامتها تنها زمانی رخ میدهند که مواد غذایی حاوی فروکتوز مصرف شوند. مصرف زیاد غذاهای حاوی فروکتوز اغلب باعث هیپوگلیسمی، بزرگی کبد، تحریکپذیری، خفگی، حمله عصبی و اختلال در عملکرد توبولهای پروکسیمال میشود. اختلال عملکرد کبد موجب طولانی شدن زمان انعقاد، افزایش ترانسآمینازها، افزایش بیلیروبین و کاهش آلبومین خون میشود.
تجمع فروکتوز 1- فسفات موجب مهار آلوستریک فسفریلاز کبدی (مهار گلیکوژنولیز) و باعث ایجاد هیپوگلیسمی ناشی از فروکتوز علیرغم وجود ذخایر زیاد گلیکوژن میشود. تحمیل حاد فروکتوز به کبد (تزریق وریدی، دریافت غذایی بسیار زیاد) موجب احتباس فسفات معدنی در فروکتوز 1- فسفات و افت ساخت ATP میشود. با اینکار، مهار آنزیمهای تجزیه نوکلئوتیدهای آدنینی که توسط ATP انجام میشد، برطرف شده و با تسریع در ساخت اسیداوریک، باعث ایجاد هیپراوریسمی میشود.
تشخیص با حدس بالینی و متعاقب آن توسط آزمایش تحمل فروکتوز داخل وریدی انجام میشود. تشخیص قطعی با اندازهگیری فعالیت آلدولاز B در نمونه بیوپسی کبد امکانپذیر است. درمان این اختلال، مصرف رژیمهای حاوی فروکتوز و سوربیتول کم میباشد.
کمبود آنزیم فروکتوز 1 و 6- بیسفسفاتاز:
یک اختلال اتوزومی مغلوب بوده که در نتیجه کمبود فروکتوز 1 و 6- بیسفسفات رخ میدهد. این آنزیم در تبدیل فروکتوز 1 و 6- بیسفسفات به فروکتوز 6- فسفات نقش دارد. در نتیجه کمبود فروکتوز 1 و 6- بیسفسفات، روند گلوکونئوژنز مختل میشود. شرایط حاد مانند هیپوگلیسمی، افزایش تهویه ریوی، اسیدوز لاکتیک، تشنج و کوما میتوانند بیمار را تحتتأثیر قرار دهند. تجمع فروکتوز 1 و 6- بیسفسفات موجب مهار آلوستریک فسفریلاز کبدی (مهار گلیکوژنولیز) و باعث ایجاد هیپوگلیسمی ناشی از فروکتوز علیرغم وجود ذخایر زیاد گلیکوژن میشود.
تشخیص این اختلال باید از طریق تأیید کمبود آنزیم در نمونههای بیوپسی کبد و یا روده، صورت گیرد. در زمان بروز حملات حاد، درمان هیپوگلیسمی و اسیدوز لاکتیک باید توسط تجویز مایعات داخل رگی انجام شود. گرسنگی طولانی که موجب تخلیه ذخایر گلیکوژن کبد میشود، سبب بروز علائم در این بیماران خواهد شد. این بیماران به محدودیت در مصرف فروکتوز و سوربیتول رژیم غذایی نیاز دارند.
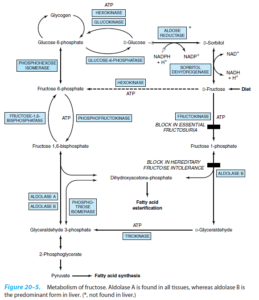
فروکتوز و سوربیتول در چشم انسان نیز یافت میشوند. افزایش غلظت فروکتوز و سوربیتول ممکن است در پاتوژنز آبمروارید (کاتاراکت) دیابتی نقش داشته باشد. مسیر سوربیتول (پلیول) (در کبد نیست)، مسئول ساخت فروکتوز از گلوکز است که با تجویز بازدارندههای آلدوز ردوکتاز ممکن است بتوان این مسیر را مهار نمود. در تجویز وریدی، سوربیتول، بیشتر به فروکتوز (تا گلوکز) تبدیل میشود. مصرف سوربیتول از راه دهان موجب گریز قسمت زیادی از آن از جذب رودهای و تخمیر به استات و H2 توسط باکتریهای روده میشود. شیرینکنندههای حاوی سوربیتول ممکن است باعث درد شکمی (عدم تحمل سوربیتول) شوند.
اختلالات ناشی از نقص در متابولیسمی گالاکتوز:

کمبود گالاکتوکیناز:
در کمبود گالاکتوکیناز، گالاکتوز به گالاکتوز 1- فسفات تبدیل نشده که منجر به ایجاد آبمروارید (کاتاراکت) میشود. تومورهای کاذب نخاع از دیگر تظاهرات این بیماری هستند. افزایش سطح گالاکتوز خون و فعالیت طبیعی اوریدیل ترانسفراز، فقدان گالاکتوکیناز در گلبولهای قرمز را اثبات مینماید. رژیم غذایی فاقد گالاکتوز در نوزادان میتواند موجب بهبودی کاتاراکت شود.
گالاکتوز 1- فسفات اوریدیل ترانسفراز:
کمبود گالاکتوز 1- فسفات اوریدیل ترانسفراز که یک بیماری ارثی اتوزوم مغلوب است، باعث ایجاد گالاکتوزومی کلاسیک میشود. اغلب جهشهای رایج ژن گالاکتوز 1- فسفات اوریدیل ترانسفراز، جهش Q188R در کروموزوم 9 میباشد. کمبود این آنزیم باعث ایجاد علائمی از قبیل هیپوگلیسمی، استفراغ، اسهال، تحریکپذیری و مشکلات تغذیهای و رشدی میشود. کودکان ممکن است یرقان، بزرگی کبد و کوفتگی داشته باشند. آزمایشهای تشخیصی اولیه، نشاندهنده وجود هیپربیلیروبینمی، افزایش ترانسآمینازهای کبدی، اسیدوز متابولیک، گالاکتوزومی، گلیکوزوری، هیپوگلیسمی و مشکلات انعقادی میباشند. این بیماران در معرض خطر پیشرفت ادم نخاع هستند. خونریزی زجاجیه و عفونتهای اشرشیاکلی نیز در برخی از این بیماران مشاهده شده است.
تجمع گالاکتوز 1- فسفات موجب کاهش فسفات معدنی کبد میشود. اشکال طولانیمدت کمبود گالاکتوز 1- فسفات اوریدیل ترانسفراز شامل آسیب به ادراک و شناخت، اختلال تخمدان در خانمها و بیماری آتاکسی نورولوژیک میشود. برای غربالگری گالاکتوزومی در نوزادان از روشهای سنجش میکروبیولوژیک و فلورومتریک استفاده میشود. برای تعیین فعالیت گالاکتوز 1- فسفات اوریدیل ترانسفراز از آزمایش غربالگری فلوروسنت نقطهای (تست بوتلر؛ Beutler test) استفاده میشود. یک آزمایش غیرمعمول برای تشخیص، تأیید بیوشیمیایی و ملکولی میباشد. سنجشهای کمی ممکن است متعاقب انتقال خون باعث ایجاد نتایج منفی کاذب شوند. یک فرم خفیف گالاکتوزومی (Duarte variant) شناخته شده که به دلیل جهش ژن گالاکتوز 1- فسفات اوریدیل ترانسفراز بوده و بهوسیله کاهش فعالیت آنزیم در گلبولهای قرمز شناسایی میشود که بهطور معمول، از نظر بالینی معنیدار نیست. درمان شامل محدود کردن گالاکتوز رژیم غذایی میباشد.
اوریدین دی فسفو گالاکتوز 4- اپیمراز:
فرم خوشخیم آن با کمبود آنزیم منحصراً در گلبولهای قرمز و گلبولهای سفید مرتبط میباشد. این افراد سالم بوده و نیازی به درمان ندارند. در فرم شدید بیماری، یافتههای بالینی شبیه به کمبود گالاکتوکیناز بوده و علاوه بر آنها، هیپوتونی و احساس ناشنوایی نیز ممکن است وجود داشته باشد. درمان این بیماری از طریق مصرف رژیم غذایی حاوی مقادیر پایین گالاکتوز امکانپذیر است.
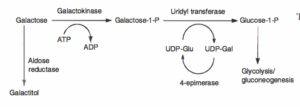
افزایش غلظت پلاسمایی گالاکتوز و احیاء آن توسط آلدوز ردوکتاز در چشم منجر به تشکیل گالاکتیتول و ایجاد آبمروارید میشود.
بیماریهای ذخیرهای گلیکوژن (Glycogen Storage Disease; GSD)
اصطلاح بیماری انباشته شدن گلیکوژن (Glycogen Storage Disease; GSD) برای یک گروه از ناهنجاریهای موروثی به کار برده میشود که با انباشته شدن مقادیر غیرطبیعی و یا انواع غیرطبیعی گلیکوژن در بافتها همراه است.
از ویژگیهای کلی این دسته از بیماریها میتوان به موارد زیر اشاره نمود:
- نتیجه فقدان آنزیمهای مسئول سنتز و یا تجزیه گلیکوژن میباشند.
- کمیت یا کیفیت گلیکوژن در این اختلالات دچار اختلال میشود.
- بیشترین تأثیر اختلال در متابولیسم گلیکوژن بر کبد و عضله میباشد، زیرا این بافتها حاوی گلیکوژن بالایی هستند.
| خصوصیات | نقص آنزیمی | بیماری | نوع |
| تجمع گلیکوژن در کبد و توبول کلیه، هیپوگلیسمی، اسیدوز لاکتیک، هیپرلیپیدمی | گلوکز 6- فسفاتاز | فون ژیرکه (Von Gierke) | l |
| تجمع گلیکوژن در لیزوزوم ها، کشنده به دلیل نارسایی قلبی | آلفا- گلوکوزیداز | پمپه (Pompes disease) | II |
| تجمع یک پلی ساکارید شاخهدار غیرمعمول (دکسترینوز محدود)، هیپوگلیسمی | شاخه شکن | کری- فوربس
(Coris or Forbes) |
III |
| تجمع یک پلی ساکارید با تعداد شاخههای کم (آمیلوپکتینوز)، هیپوگلیسمی | شاخه ساز | آندرسن (Andersen) | IV |
| تجمع گلیکوژن در عضله، اختلال در فعالیت عضلانی | فسفریلاز عضلانی | مک آردل (McArdel) | V |
| تجمع گلیکوژن در کبد، هیپوگلیسمی | فسفریلاز کبدی | هرس (Hers) | VI |
| کمبود در عضله و گلبولهای قرمز، اختلال فعالیت عضلانی و احتمالاً کمخونی | فسفوفروکتوکیناز | تاروی (Tarui) | VII |
فون ژیرکه (Von Gierke’s disease) (GSD-I):
این بیماری ناشی از نقص در آنزیم گلوکز 6- فسفاتاز بوده و شایعترین بیماری از دستهی بیماریهای GSD میباشد. این بیماری موجب اختلال کبد در تولید گلوکز از گلیکوژن و گلوکونئوژنز شده و ازاینرو میتواند باعث ایجاد هیپوگلیسمی شدید و بزرگی کبد شود. لاکتیک اسیدوزیس، هیپرلیپیدمی و هیپراوریسمی نیز از نشانههای این بیماری میباشند. هیپوگلیسمی ناشی از GSD-I بهعنوان هیپوگلیسیمی ناشتایی (fasting) یا هیپوگلیسیمی پس از جذب (post-absorptive) نامیده میشود که به معنی وقوع هیپوگلیسمی پس از جذب کامل غذا (معمولاً 4 ساعت بعد) میباشد. کمبود گلوکز 6- فسفاتاز موجب افزایش ریسک ابتلا به آدنوکارسینومای کبدی (Hepatic adenocarcinoma) میشود. درمان این بیماری، مصرف مداوم و پیوسته کربوهیدراتها میباشد.
پمپه (Pompes disease) (GSD-II):
این بیماری ناشی از نقص در آنزیم آلفا- گلوکوزیداز بوده و موجب تجمع گلیکوژن در لیزوزومها میشود. تجمع گلیکوژن موجب ضعف پیشرونده عضلانی در سراسر بدن شده و بافتهای مختلفی بهویژه قلب، عضلات اسکلتی، کبد و سیستم عصبی را تحتتأثیر قرار میدهد. تشخیص از طریق رادیوگرافی قفسه سینه (Chest X ray)، الکتروکاردیوگرام و اکوکاردیوگرافی میباشد. افزایش سطح کرآتین کیناز سرمی (تقریباً یک افزایش 10 برابری) همراه با افزایش کمتری در آلدولاز، ترانسآمینازها و لاکتات دهیدروژناز نیز مشاهده میشود. تشخیص ممکن است که از طریق تخمین فعالیت آلفا-گلوکوزیداز در بیوپسی پوست، عضله و یا در گلبولهای سفید داده شود.
کری- فوربسCori’s or Forbes disease) (GSD-III)):
این بیماری ناشی از نقص در آنزیم شاخهشکن بوده و موجب تجمع یک گلیکوژن غیرطبیعی در کبد و عضلات میشود. هیپوگلیسمی، هپاتومگالی، هیپوتونی و … از علائم این بیماری هستند. درمان از طریق مصرف رژیم غذایی حاوی مقادیر زیاد پروتئین به منظور تسهیل روند گلوکونئوژنز میباشد.
آندرسن (Andersen) (GSD-IV):
این بیماری ناشی از نقص در آنزیم شاخه ساز گلیکوژن بوده که منجر به ایجاد یک زنجیره بسیار طویل بدون شاخه از گلوکز میشود. این زنجیره طولانی بدون شاخه از گلوکز دارای حلالیت پایینی بوده و منجر به رسوب گلیکوژن در کبد و ایجاد سیروز میشود.
مک آردل (McArdel) (GSD-V):
این بیماری ناشی از نقص در آنزیم فسفریلاز عضلانی میباشد. علائم این بیماری شامل عدم تحمل ورزش همراه با درد عضلانی، خستگی، کرامپهای دردناک و میوگلوبینوری میباشد. در این بیماری، بیماران ممکن است پدیده هوای ثانویه (Second wind phenomenon) از خود نشان دهند؛ بدین معنی که فرد، ورزشهای هوازی از قبیل پیادهروی را پس از مدت زمان 10 دقیقه، بهتر تحمل مینماید که احتمالاً به دلیل افزایش گردش خون و توانایی بدن در استفاده از منابع انرژی جایگزین از قبیل اسیدهای چرب و پروتئینها میباشد.
در بیوپسی عضله این بیماران، میوفسفریلاز مشاهده نمیشود. تعیین ردیف ژن PYGM نیز ممکن است که به منظور تشخیص این بیماری انجام شود. افزایش متوسط سطح کرآتین کیناز در 90% این بیماران مشاهده میشود. تجویز ویتامین B6 خوراکی موجب کاهش خستگی در این افراد میشود. درمان خاصی برای این بیماری وجود ندارد. ورزشهای هوازی و مصرف رژیم غذایی سرشار از پروتئین در این افراد ممکن است کمککننده باشد.
هرس (Her’s disease) (GSD-VI):
این بیماری ناشی از نقص در آنزیم فسفریلاز کبدی میباشد. این بیماری تقریباً 30% موارد بیماریهای ذخیرهای گلیکوژن را به خود اختصاص میدهد. هپاتومگالی و تأخیر در رشد در دوران نوزادی از علائم این بیماری میباشد. هیپوگلیسیمی متوسط، هیپرلیپیدمی و هیپرکتوزیز نیز ممکن است مشاهده شوند. سطح اسیدلاکتیک و اسید اوریک ممکن است که طبیعی باشند. در طی ناشتایی ممکن است که لاکتیک اسیدوزیس رخ دهد.
تاروی (Tarui’s disease) (GSD-VII):
این بیماری ناشی از نقص در آنزیم فسفوفروکتوکیناز میباشد. در این بیماری، کمبود زیرواحد M آنزیم فسفوفروکتوکیناز موجب اختلال در توانایی برخی سلولها از قبیل گلبولهای قرمز و سلولهای عضلانی در استفاده از کربوهیدراتها بهعنوان منبع انرژی میشود. این بیماری با گرفتگیهای عضلانی متعاقب ورزش، ضعف، میوگلوبینوری و آنمی همولیتیک همراه میباشد. هیپراوریسمی نیز در این بیماری، شایع میباشد. تشخیص میتواند از طریق بیوپسی عضله و حضور مقادیر زیاد گلیکوژن در آن و همچنین بررسی سطح آنزیم فسفوفروکتوکیناز تأیید شود. همچنین میتوان از آزمایشهای DNA به منظور تعیین موتاسیون در ژن فسفوفروکتوکیناز استفاده نمود.
منابع:
1-Tietz Textbook of Clinical Chemistry and Molecular Diadnosis. 2006; 4th Edition.
2- Henrys Clinical Diagnosis and Management by Laboratory Methods. 2007; 21st Edition.
3 – Wendy Aineson and Jean Brickell. Clinical Chemistry; A Laboratory Perspective. 2007; 1st Edition.
4- Arneson W, Brickell J. Clinical chemistry; a laboratory perspective. 2007.
5- Pagana KD and Pagana TJ. Diagnostic and laboratory test refrence. 2005; 7th Edition.
6- Van Leeuwen AM, Kranpitz TR and Smith L. Laboratory and diagnostic tests with nursing implications. 2006; 2nd Edition.
7- Wilson DD. Manual of laboratory and diagnostic tests. 2008.
گلوکز-6- فسفات دهیدروژناز(در تب جدید مرورگر باز می شود )
https://www.ncbi.nlm.nih.gov/books/NBK560599/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام