مروری بر لنفوم بورکیت
الهام عبدالهی1،2، امیرعباس ممتازی بروجنی3، فتانه توسلیان 4دانشجوی دکترای تخصصی ایمونولوژی پزشکی
1: کمیته تحقیقات دانشجویی، دانشگاه علوم پزشکی مشهد
2: دانشجوی دکترای تخصصی بیوتکنولوژی پزشکی مشهد
3: دانشجوی دکترای تخصصی ایمونولوژی پزشکی، تهران
چکیده: لنفوم بورکیت نوعی سرطان سیستم لنفاوی و نئوپلازی غیرطبیعی است که بیشتر استخوانهای فک و گونه را درگیر میکند. این بیماری در اواخر دهه 1950 توسط دنیس بورکیت توصیف شد. مطالعات سیتوژنیک نشان داده است که در اغلب بیماران (حدود 90%) انکوژن c-myc از کروموزوم 8 با ژن زنجیره سنگین ایمونوگلوبولین H بر روی کروموزوم 14 جابهجا شده است. در موارد نادرتر، آنکوژن myc با نواحی از کروموزوم 2 و 22 جابهجا میشود که این نواحی ژنهای کدکننده زنجیرههای سبک کاپا و لامبدا آنتیبادی را کد میکنند. در اثر این جابهجایی ژن myc تحت کنترل پروموتر ژن ایمونوگلوبولین قرار میگیرد و بیان آن تا حد 10 برابر و یا بیشتر افزایش پیدا میکند. علاوه بر پروتوانکوژن C-myc عوامل دیگری نیز در پاتوژنز لنفوم بورکیت نقش دارند. در این مطالعه مروری سعی بر این است تا جنبههای مختلف، بهویژه مکانیزم مولکولی جدید مطرحشده در ایجاد لنفوم بورکیت موردبررسی قرار گیرد.
واژگان کلیدی: نئوپلازی، لنفوم بورکیت، انکوژن c- myc
مقدمه
ویژگی بسیاری از بدخیمیها با ریشهی خونی، عدم تنظیم فعالیت ژنهای دخیل در رشد سلولی، بقا و تمایزاست، یکی از عوامل مهم ایجاد این اختلالات در سطح سیتوژنتیک جابهجاییهای کروموزومی میباشد. در بسیاری از موارد عناصر تنظیمی مربوط به ژنهای ایمونوگلوبینها و رسپتور لنفوسیتهای T در مجاورت ژنهای پروتوانکوژنها قرار میگیرند که منجر به افزایش بیان این ژنها میشود. لنفوم بورکیت یکی از نمونههای بارز بدخیمیهای لنفوسیتهای B است که شیوع آن در جمعیت انسانی بهسرعت در حال رشد است. از نشانههای آن میتوان به متورم شدن گرههای لنفاوی در گردن، زیر فک، کشاله ران و زیر بغل اشاره کرد (1). لنفوم بورکیت برای اولین بار توسط دنیس بورکیت در سال 1958 معرفی شد. (2-4).
لنفوم بورکیت به سه شکل بومی، پراکنده و همراه با ویروس نقص ایمنی انسانی دیده میشوند (جدول 1). (1, 5)
لنفوم بورکیت بومی
این نوع لنفوم در مناطق گرم و مرطوب مانند خاورمیانه، شمال افریقا (که در آن شیوع بیماری مالاریا بالاست) و مناطقی از امریکای جنوبی شیوع بالایی دارد (6, 7). مشخصهی این نوع لنفوم درگیری فک و سایر استخوانهای صورت است. در این نوع لنفوم احتمال درگیری سیستم عصبی مرکزی و مغز استخوان بین 12 تا 22 درصد میباشد(8). تقریباً تمام موارد نوع بومی با عفونت ویروس اپشتاین بار (EBV) همراه است(9) .
لنفوم بورکیت پراکنده
این نوع لنفوم بیشتر در کودکان جوامع پیشرفته مانند کشورهای امریکای شمالی و اروپایی مشاهده شده است. در این نوع لنفوم درگیری غدد لنفاوی در بالغین بیشتر از کودکان است(10). برخلاف نوع بومی که فک را بیشتر درگیر میکند، در نوع پراکنده درگیری فک بهندرت مشاهده میشود(11). این نوع لنفوم از نوع تومورهای مهاجم است که میتواند سیستم عصبی مرکزی و مغز استخوان را بهشدت تحت تأثیر قرار دهد(12). درگیری روده بهخصوص ناحیه ایلئوم و غدد لنفاوی در روده در این نوع لنفوم بیشتر است. همچنین تخمدان، کلیه، پانکراس، کبد، بافت سینه در زنان و مغز استخوان نیز تحت تأثیر این نوع لنفوم قرار میگیرند(13, 14).
لنفوم بورکیت همراه با عفونت ویروس نقص ایمنی انسان (HIV)
لنفوم بورکیت همراه با عفونت ویروس نقص ایمنی انسان (HIV) در افراد مبتلا به ویروس HIV مشاهده میشود. همچنین در افرادی که پیوند عضو دریافت نمودهاند و تحت درمان با داروهای سرکوب سیستم ایمنی قرار گرفتهاند(15، 16) و در افراد مبتلا به نقص ایمنی مادرزادی نیز مشاهده میشود(17). بهطورکلی حدود 30 درصد از موارد لنفوم بورکیت مرتبط با ویروس HIV است و احتمال ابتلا افراد مبتلا به این ویروس به لنفوم بورکیت نسبت به افراد معمولی هزار برابر بیشتر است (17). احتمال عفونت با ویروس اپشتاین بار در این نوع لنفوم 40 تا 50 درصد، وابسته به شرایط مختلف دموگرافیک است (18).
ویژگی مشترک این سه نوع لنفوم این است که سلولهای لنفومایی از تمایز سلولهای B در مناطق زایگر بافتهای لنفاوی ایجاد میشوند. همچنین جهشهای سوماتیک با میزان بالا در مناطق متغیر (V) زنجیره سنگین ایمونوگلوبین رسپتور لنفوسیتهای B در این مناطق بیشتر از میزان معمول در رسپتور سلولهای B معمولی است(18).
جدول 1: ویژگی آسیبشناسی بالینی در لنفوم بورکیت

مکانیزم مولکولی ایجاد لنفوم بورکیت
پروتوانکوژن c-myc بهطورمعمول در ایجاد سرطانهای انسانی دخیل است. این پروتئین در سیگنالرسانی سلولی، اپوپتوز، چسبندگی سلولها، سنتز پروتئین، تنظیم فرآیندهای سلولی مثل تمایز، تکثیر از طریق افزایش سیکلین D، فاکتور E2F و کیناز 4 وابسته به سیکلین (CDK4) (کمپلکسهای سیکلین /CDK نقاط مختلف سیکل سلولی را تنظیم میکنند CDK4 باعث پیشرفت از فاز G میشود(19) متابولیسم، آپوپتوز، تعمیر و حفظ تلومر مشارکت دارد و فعالیت آن بهشدت در سطح رونویسی و پس از رونویسی کنترل میشود. در لنفوم بورکیت کنترل پس از رونویسی غالب است. پروتوانکوژن c-myc منجر به سرکوب ژنهای مهار تقسیم سلولی مانند p57،P15، P21,P27,GADD45,GADD34,GADD153) میشود(20،21). C-myc به همراه پروتئینهای Bax ( pro-apoptotic Bcl-2-associated X protein) منجر به افزایش فرایند اپوپتوز میشود (22).
c-myc یک پروتئین 64 کیلو دالتونی است که متعلق به خانواده فاکتور رونویسی هستهای زیپ لوسینی هلیکس- مارپیچ- هلیکس است(23, 24).
قسمت کربوکسی این پروتئین شامل 340 اسیدآمینه با ساختار هلیکس- مارپیچ- هلیکس است. این قسمت میتواند به DNA متصل شود. پس از اتصال به DNA، میتواند با فاکتور X همراه با پروتئین myc (MAX) هترودایمر شود. تشکیل این کمپلکس برای ایجاد فرم فعال myc که قادر به اتصال به توالی جعبه تقویتکننده (E-Box) در DNA هدف باشد ضروری است که نتیجه آن افزایش رونویسی DNA هدف میباشد (25،26).
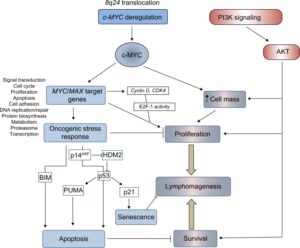
شکل 1: مسیرهای تنظیمکننده تکثیر، بقا و مرگ سلولی در لنفوم بورکیت
لنفوم بورکیت به علت ایجاد یک جابجایی کروموزومی از نوع متعادل و دوطرفه ایجاد میشود. جابهجایی بین ژنهای مربوط به ایمونوگلوبولین در کروموزومهای 14،2،22 و c-myc در کروموزوم 8 رخ میدهد(27). در 80% موارد لنفوم بورکیت، جابجایی c-myc به لوکوس ایمونوگلوبولین H است که منجر به تشکیل (q24:q32 )( 8:14) میشود. در 15% موارد جابجایی به لوکوس کاپا در کروموزوم 2q11 و در 5% موارد به لوکوس لامبدا در کروموزوم 22q11 میباشد. درنتیجهی این جابهجاییها ژن C-myc ازلحاظ رونویسی بهشدت فعال میشود .در این حالت پروموتور یک ( P1) ژن C-myc بیشتر از پروموتور 2 (P2، که در حالت عادی فعال است) فعال میشود. که بهعنوان تغییر پروموتوری (promotor shifting ) شناخته میشود (27, 28).
موقعیت شکستگی در ژن زنجیره سنگین ایمونوگلوبولین و c-myc
موقعیت شکستگی کروموزوم در ژن c-myc و ژن زنجیره سنگین ایمونوگلوبولین بسیار متفاوت است، در کروموزوم ژن c-myc جابهجایی در ناحیه 8q32 رخ میدهد که وسیلهی کاریوتایپ کروموزومهای متافازی یا وسیلهی هیبریداسیون درجا تشخیص داده میشود (شکل 2 و 3) (29).
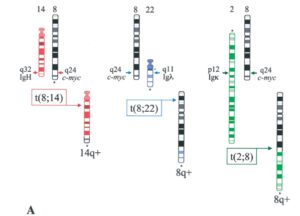
شکل 2: جابهجایی بین کروموزوم 8،14،22،2
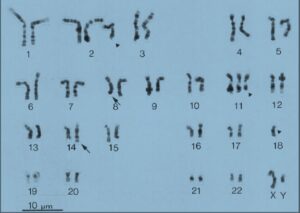
شکل 3: کاریوتایپ سلولی با جابهجایی (14: 8)
در لنفوم بورکیت نوع بومی شکستگی روی کروموزوم 8 در پشت ‘5 اگزون شماره 1 c-myc رخ میدهد (شکل B4)، درحالیکه شکستگی روی کروموزوم 14 در ناحیه (IgH) (JH) اتفاق میافتد.
در لنفوم بورکیت پراکنده و ویروس نقص ایمنی انسانی شکستگی (14: 8) باعث حذف ناحیه بین اگزون 1 و 2 c-myc روی کروموزوم 8 و درون ناحیه IgHSμ کروموزوم 14 میشود (شکل c4) (29).
در نوع دوم جابهجایی لوکوس c-myc به لوکوس ایمونوگلوبولین کاپا روی کروموزوم 2 یا لوکوس ایمونوگلوبولین لامبدا روی کروموزوم 22 وصل میشود. شکستگی روی کروموزوم 8 پشت اگزون c-myc است (شکل 4- E و D) شکستگی روی کروموزوم 2 و 22 در ‘5 ژن کاپا و لامبدا رخ میدهد(29).
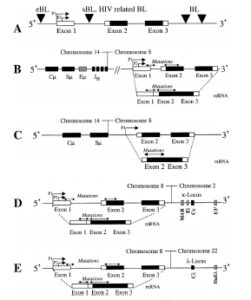
شکل 4: موقعیت شکستگی روی C-Myc و Ig
تغییراتی در بیان ژن c-myc با ایجاد جابجایی ایجاد میشود:
- بیان این ژن به میزان بالایی افزایش مییابد درحالیکه در حالت معمولی این ژن ازلحاظ رونویسی غیرفعال است و یا رونویسی و بیان آن به میزان اندک است(30،31) در این حالت نیمهعمر mRNAی ایجادشده بسیار کوتاه است (32).
- پروموتور تنظیمکننده رونویسی در این حالت پروموتور 1 است درحالیکه سنتز حدود 80 تا 90 درصد از RNAی مربوط به ژن c-myc در حالت طبیعی توسط پروموتور 2 تنظیم میشود (33).
- ازلحاظ ساختاری ژن C-myc در ناحیه اولین اگزون یا اولین اینترون دچار تغییر میشود(33).
- تنظیم طول RNA مربوط به ژن myc مختل میشود (33).
- بیان ژن myc در حالت جابجایی توسط سدیم بوتیرات کاهش مییابد (33).
در مطالعات انجام شده مکانیزمهای مختلفی در افزایش بیان ژن در حالت ایجاد جابجایی پیشنهاد شده است:
در مطالعهای نشان داده شد که در جابهجایی شایع (t(8,14 وجود عناصر تقویتکننده در نواحی اینترونی در لوکوس ژنی زنجیره سنگین µ و تقویتکننده لوکوس ژنی Cα برای افزایش بیان ژن c-myc ضروری است(31).
در مطالعه دیگری نشان داده شد که در لوکوس ژنی K در کروموزوم 2، عنصر تقویتکننده در ناحیه اینترونی در سر ‘3 ژن K (Ei3’) منجر به افزایش بیان ژن c-myc میشود که منجر به تغییر پروموتوری و همچین مختل شدن تنظیم طول RNA میشود (34)
یافتهها در مورد مکانیسم مؤثر در افزایش بیان ژن c-myc در لوکوس ژنی λ در کروموزوم 22 اندک است در مطالعهای نشان داده شد که توالی تقویتکننده در ناحیه 3’ لوکوس λ ناحیه کاندید در افزایش بیان ژن c-myc میباشد(35).
علاوه بر این چندین جایگاه بسیار حساس hyper sensitive)) به DNAase I به نام HSS که مابین ژن Cλ و توالی تقویتکننده λ وجود دارد و هنگامیکه این ناحیه در مجاورت ژن c-myc در کروموزوم 8 پس از ایجاد جابهجایی t(8,22) قرار میگیرد، رونویسی ژن c-myc افزایش مییابد (36).
در مطالعهای تأثیر نواحی خاصی از توالی تقویتکننده به نام HUE λ (که جزو عناصر تنظیمی ژن است) و به طول 12bp در لوکوس لامبدا که طی جابجایی در مجاورت ژن C-myc قرار میگیرد مورد بررسی قرار گرفت و نشان داده شد که این ناحیه منجر به تغییر پروموتور از P2 به P1 شد (37).
پروتئین C-myc پس از فعال شدن میتواند منجر به فعال شدن سایر مسیرهای سیگنالرسانی مانند مسیر فسفواینوزیتید 3 کیناز (PI3K) شود. علاوه براین در ژن myc نیز ممکن است جهش ایجاد شود. انتهای امینی پروتئین myc شامل دو توالی حفظشده به نامهای BOX1 و BOX2 میباشد(38). موتاسیون در توالی BOX2 منجر به افزایش اپوپتوز ایجادشده توسط پروتئین c-myc میشود(39). توالی Box1 دارای مکانهایی جهت فسفریله شدن است که در پروتئولیز با واسطه c-myc بهوسیله پروتئازوم-یوبیکوئیتین دخیل است (40). این دو ناحیه در حدود 20 درصد از موارد لنفوم بورکیت دچار جهش میشوند . این جهشها بهویژه در نواحی داغ (Hot spot) در تبدیل اسیدآمینه ترئونین مکانهای 39،57،78 به اسیدآمینه الانین اتفاق میافتد که نتیجتاً منجر به یوبیکوئیتانسیون ناقص،کاهش تجزیه پروتئازومی و افزایش پایداری پروتئین c-myc میشود(41-43). انواع جهشهای دخیل در ایجاد لنفوم بورکیت در شکل 5 نشان داده شده است.
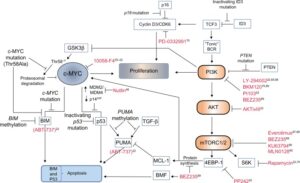
شکل 5: نقش جهشها در ایجاد لنفوم بورکیت و روشهای درمانی مبتنی بر آنها
نقش ژن P53 در پاتوژنز لنفوم بورکیت
ژن P53 که در سال 1993 بنام مولکول سال و ژن نگهبان شناخته شد بهطور طبیعی تقسیم و رشد سلول را تحت نظر کامل دارد. هنگامیکه این ژن موتاسیون پیدا میکند باعث تولید یک پروتئین غیرمعمولی میشود که نهفقط به اعمال طبیعی خود جامه عمل نمیپوشاند بلکه همه ژنهایی که تحت فرماندهی این پروتئین انجاموظیفه میکردند طغیان خواهند کرد و یک سری از روابط مولکولی و بیولوژیکی تقسیم سلولی از مسیر طبیعی خود خارج میشود و سلول بهسوی سرطانی شدن پیشروی میکند. موتاسیون ژن P53 در بیش از 60 درصد بافتهای سرطانی دیده میشود(44). همچنین موتاسیون این ژن در حدود 30 تا40 درصد از موارد لنفوم بورکیت مشاهده میشود(45،46) و حذف در ناحیه 6q در 30 درصد از موارد مشاهده شده است (47).
نقش مسیر PI3k در ایجاد لنفوم بورکیت: افزایش فعالیت ژن C-myc بهتنهایی در ایجاد لنفوم بورکیت دخیل نیست. تأثیرات همزمان مسیر C-myc و PI3K برای نخستین بار توسط ساندر و همکاران موردبررسی قرار گرفت. نقش PI3K در کنار عامل c-myc در افزایش بقای سلولهای لنفومایی در بسیاری از مطالعات موردبررسی قرارگرفته است (48-51).
همچنین نقش مسیر PI3K در تکثیر و بقا سلولهای لنفومایی در مطالعه دیگری نشان داده شد (52).
در مطالعهای نشان داده شد که جهشهای شایع در 70 درصد از موارد لنفوم بورکیت باعث افزایش مسیر سیگنالرسانی PI3K میشود(53).
در مطالعهای جهشهای معمول در ژن PI3KR1 مشاهده شد. محصول این ژن شامل پروتئینی هترودایمر متشکل از یک زیر واحد کاتالیتیک (p110α,p110B یا p110δ) و یک زیر واحد تنظیمی (p85α,p55α,p50α) است که هردو زیر واحد از یک ژن PI3KR1 حاصل شدهاند. زیر واحد تنظیمی P55α پس از عفونت سلولهای B لنفومایی در لنفوم بورکیت به ویروس اپشتاین بار، برای بقا و تکثیر سلولهای لنفوبلاستوئیدی موردنیاز است (54).
همانند سایر سرطانها، سیگنال رسانی مسیر PI3K میتواند در پایداری پروتئین myc از طریق تنظیم فعالیت گلیکوژنسنتازکیناز 3B (GSK3B)، کینازی که در پاییندست PI3K فعال میشود، مؤثر باشد(55).
روشهای درمانی مبتنی بر مسیر PI3K
مسیر PI3k بسیاری از عملکردهای سلولی ازجمله تکثیر سلولی و اپوپتوز را کنترل میکند. PI3k این کنترل را از طریق فعال کردن عوامل پاییندست خود انجام میدهد که معروفترین آنها یک سرینترهاونینکیناز به نام AKt میباشد (نام دیگر AKt پروتئین کیناز B است) (56).
در مطالعات مختلف نقش پروتئینکیناز B که بهعنوان AKT شناخته میشود و mTOR (پروتئین هدف راپامایسین در پستانداران) فعالشده در پاییندست مسیر PI3K در پاتوژنز لنفوم بورکیت و درمان آن موردتوجه قرارگرفته است (57).
در مطالعهای نشان داده شد که اپوپتوز در سلولهای لنفومایی بهوسیلهی مهارکننده مسیر PI3K
(Ly-294002) و مهارکننده PI3K\mTOR (PI103) القا میشود (58).
مهارکنندههای AKT (َAKTi-V3) بهتنهایی نیز منجر به اپوپتوز در سلولهای لنفومایی میشوند که نشانگر این است که سیگنالرسانی PI3K بهواسطه AKT بر بقای سلولهای لنفومایی در لنفوم بورکیت حائز اهمیت است (59).
ویژگیهای بافتشناسی لنفوم بورکیت
در بیماران مبتلا به لنفوم بورکیت سلولها در بافت خون و مغز استخوان دارای اندازه متوسط، هسته گرد، چندین هستک چسبیده به غشا هستند. سیتوپلاسم بازوفیلی و اغلب حاوی واکوئلهایی از جنس چربی هستند (20).
از دیگر مشخصات بافتی لنفوم بورکیت میتوان به حضور سلولهای آپوپتوزی متعدد درون ماکروفاژهای فاگوسیت کننده و همچنین وجود سلولهای بسیاری با مارکرCD10/CD20/، Igm که در زیر میکروسکوپ ظاهری شبیه آسمان پرستاره به خود میگیرند اشاره کرد. این ویژگی بهوسیله رنگآمیزی با نشانگرهای خاص مانند ki-67 مشخص میشود، در این شکل همچنین نشان داده شده بیش از 95% سلولهای توموری در حال تکثیرند (شکل 6) (19).
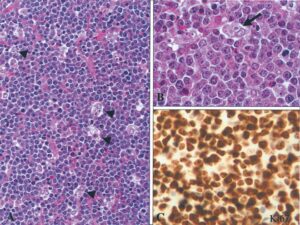
شکل 6: A،B سلولهای ماکروفاژی فاگوسیت کننده، C نشانگر ki-67
خصوصیات ایمونوفنوتیپیک لنفوم بورکیت
سلولهای لنفومایی B در لنفوم بورکیت بهطورمعمول Igm سطحی، CD19، CD20،CD22 ،CD10 ،BCL-6 و CD79a را بیان میکنند و مارکرهای CD5، CD23 وBCL-2 را بیان نمیکنند (BCL-2 در لنفومهای فولیکولار بیان میشود)(60). علاوه بر این در برخی از موارد فقدان ایمونوگلوبینهای سطحی مشاهده شده است (61).
تظاهرات بالینی لنفوم بورکیت
تظاهرات بالینی لنفوم بورکیت معمولاً در مدت کوتاهی شناسایی میشوند زیرا سلولهای لنفاوی بسیار سریع تکثیر میشوند. بهاحتمالزیاد یک یا چند گره لنفاوی متورم میشود. در بسیاری از افراد با لنفوم بورکیت ممکن است رودهها نیز تحت تأثیر قرار بگیرند و ممکن است درد شکم و اسهال ایجاد شود. گاهی ممکن است این علائم با علائم درد آپاندیس اشتباه گرفته شوند. گاهی تجمع مایعات در داخل روده مشاهده میشود که آسیت نامیده میشود که ممکن است سبب انسداد و خونریزی روده بشود(2).
علائم لنفوم بورکیت ممکن است خارج از گرههای لنفاوی نیز رخ دهد بنابراین ممکن است سایر نواحی نیز درگیر شوند. علائم دیگری مانند تعریق شبانه، خستگی، علائمی مانند علائم سرماخوردگی، کاهش وزن غیرقابل توضیح نیز ممکن است وجود داشته باشد (2).
لنفوم بورکیت میتواند مغز و نخاع را نیز درگیر میسازد، در صورت درگیر شدن سیستم عصبی مرکزی علائمی مانند سردرد، تشنج، گیجی و عدم تمرکز به وجود میآید. لنفوم بورکیت ممکن است سایر اندامها مانند طحال، کلیه، کبد و سینه را نیز درگیر سازد. شایانذکر است که در کودکان با لنفوم بورکیت از نوع بومی بیشتر فک تحت تأثیر قرار میگیرد درحالیکه در نوع پراکنده لنفوم بورکیت این درگیری در کودکان کمتر دیده میشود (2).
تشخیص لنفوم بورکیت
لنفوم بورکیت معمولاً با نمونهبرداری از گرههای لنفاوی متورم یا بافتهای تحت تأثیر قرار گرفته تشخیص داده میشود. در سالهای اخیر روشهای تشخیصی بهتری برای تشخیص این بیماری گسترش یافته است. این روشها شامل، بررسی میکروسکوپی سلولها و مشاهده پروتئینهای خاص در سلول و بررسی تغییر ژنها است. در این بررسیها تقسیم سلولها و همچنین موقعیت تغییر یافته ژن Myc ارزیابی میشوند. بهمنظور تشخیص لنفوم بورکیت روشهای دیگری مانند بررسی مغز استخوان، سیتیاسکن، MRI و … نیز ممکن است انجام شود (2).
درمان لنفوم بورکیت
هنگامیکه درمانهای مورداستفاده درمان لنفوم غیر هوچکین یا لوسمی لنفوبلاستیک حاد (ALL) برای درمان لنفوم بورکیت مورداستفاده قرار گرفتند پاسخ به درمان بیماران حدود 30 تا 70 درصد بود اما میزان درمان موفق صفر تا 30 درصد بود(62-65).
داروهایی که امروزه درمان لنفوم بورکیت مورداستفاده قرار میگیرند:
BNHC-83: شامل سیکلوفسفامید، پردنیزون، متوترکسات، سیتورابین، دوکسوروبیسین، لئوسودورین میباشد (66).
BNHC86: مشابه BNHC-83 با این تفاوت که ایفوسفاماید و دگزامتازون به ترتیب جایگزین سیکلوفسفامید و پردنیزون شده است (62).
CODOX-M/IVAC: حاوی سیکلوفسفامید، وینکریستین، دوکسوروبیسین، متوترکسات با دوز بالا، ایفوسفامید، اتوپوسید، سیتورابین با دوز بالا (Ara-C) (67).
hyperCVAD به همراه ریتوکسیماب (Retuximab) یا بدون استفاده از آن: حاوی سیکلوفسفامید، وینکریستن، دوکسوروبسین و دگزامتازون است (68).
به دلیل اینکه سلولهای لنفومایی بیان کننده مارکر CD20 در سطح خود هستند و آنتیبادی منوکلونال ضد ان ریتوکسیماب است این آنتیبادی نیز در درمان لنفوم بورکیت مؤثر است (69).
مهارکنندههای آنزیم تلومراز
تلومرها توالی حفاظت شده در انتهای کروموزومهای انسانها هستند. آنزیم تلومراز در سلولهای بنیادی که قدرت تکثیری بالایی دارند جهت حفظ طول تلومرها به کار میروند زیرا در اثر تقسیم سلولی طول تلومرها کاهش مییابد و آنزیم تلومراز در بیشتر سلولهای توموری فعال ولی در سلولهای طبیعی غیرفعال است. مهار هر یک از زیرواحدهای آنزیم تلومراز هدف مناسبی در طراحی داروهای مولکولی برای درمان بدخیمیها در انسان میباشد (70). در لنفوم بورکیت افزایش فعالیت آنزیم تلومراز وجود دارد(71) لذا این راهکار درمانی برای درمان لنفوم بورکیت میتواند مفید باشد.
علاوه بر درمان دارویی، شیمیدرمانی و پرتودرمانی هم برای درمان لنفوم بورکیت مورداستفاده قرار میگیرند(29).
پیوند مغزاستخوان: تأثیر پیوند مغز استخوان اتولوگ یا آلوژن برای درمان لنفوم بورکیت مباحثه برانگیز است. اگرچه برخی از مطالعات نشان دادهاند که درمان بهوسیله پیوند سلولهای بنیادی باعث افزایش بقای بیمار میشود (72، 73) ولی این نوع درمان هنوز بهعنوان روش درمانی استاندارد مورد پذیرش قرار نگرفته است.
سایر روشهای درمانی: بهتازگی مشخص شده است که مهارکننده سروتونین (SSRI) دارای تأثیرات اپوپتوتیک بر سلولهای لنفومایی است (74, 75).
نتیجهگیری
به نظر میرسد درک مکانیسمهای مولکولی جدید دخیل در ایجاد لنفوم بورکیت میتواند افقهای درمانی جدیدی را مطرح نماید. لذا پیشنهاد میشود که انجام مطالعات آینده در این زمینه متمرکز شود
منابع:
- Mangani D, Roberti A, Rizzolio F, Giordano A. Emerging molecular networks in Burkitt’s lymphoma. Journal of cellular biochemistry. 2013;114(1):35-8.
- Linch DC. Burkitt lymphoma in adults. British journal of haematology. 2012;156(6):693-703.
- Burkitt D. A sarcoma involving the jaws in African children. British Journal of Surgery. 1958;46(197):218-23.
- Burkitt D, O’Conor GT. Malignant lymphoma in African children. I. A clinical syndrome. Cancer. 1961;14(2):258-69.
- Manolova Y, Manolov G, Kieler J, Levan A, Klein G. Genesis of the 14q+ marker in Burkitt’s lymphoma. Hereditas. 1979;90(1):5-10.
- Cardy AH, Sharp L, Little J. Burkitt’s lymphoma: A review of the epidemiology. Kuwait Medical Journal. 2001;33(4):293-306.
- Makata AM, Toriyama K, Kamidigo NO, Eto H, Itakura H. The pattern of pediatric solid malignant tumors in western Kenya, east Africa, 1979–1994: an analysis based on histopathologic study. American Journal of Tropical Medicine and Hygiene. 1996;54(5):343-7.
- Cairo MS, Sposto R, Perkins SL, Meadows AT, Hoover‐Regan ML, Anderson JR, et al. Burkitt’s and Burkitt‐like lymphoma in children and adolescents: a review of the Children’s Cancer Group Experience*. British journal of haematology. 2003;120(4):660-70.
- Magrath I. The pathogenesis of Burkitt’s lymphoma. Advances in cancer research. 1990;55:133-270.
- Boerma E, van Imhoff GW, Appel IM, Veeger NJ, Kluin PM, Kluin-Nelemans J. Gender and age-related differences in Burkitt lymphoma–epidemiological and clinical data from The Netherlands. European Journal of Cancer. 2004;40(18):2781-7.
- Sariban E, Donahue A, Magrath I. Jaw involvement in American Burkitt’s lymphoma. Cancer. 1984;53(8):1777-82.
- Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood. 2004;104(10):3009-20.
- Magrath I, Sariban E. Clinical features of Burkitt’s lymphoma in the USA. IARC scientific publications. 1985 (60):119.
- Gholam D, Bibeau F, El Weshi A, Bosq J, Ribrag V. Primary breast lymphoma. Leukemia & lymphoma. 2003;44(7):1173-8.
- Gong JZ, Stenzel TT, Bennett ER, Lagoo AS, Dunphy CH, Moore JO, et al. Burkitt lymphoma arising in organ transplant recipients: a clinicopathologic study of five cases. The American journal of surgical pathology. 2003;27(6):818-27.
- Xicoy B, Ribera J-M, Esteve J, Brunet S, Sanz M-A, Fernández-Abellán P, et al. Post-transplant Burkit t’s Leukemia or Lymphoma. Study of Five Cases Treated with Specific Intensive Therapy (PETHEMA ALL-3/97 Trial). Leukemia & lymphoma. 2003;44(9):1541-3.
- Ferry JA. Burkitt’s lymphoma: clinicopathologic features and differential diagnosis. The Oncologist. 2006;11(4):375-83.
- Crawford DH. Biology and disease associations of Epstein–Barr virus. Philosophical Transactions of the Royal Society B: Biological Sciences. 2001;356(1408):461-73.
- Kamali E, Ghaedi K, Karimi P, Kheradmand P, Tavassoli M, کمالي ا، et al. تأثيرات بيولوژيک و ضد سرطاني کورکومين (Curcumin). مجله دانشکده پزشکی اصفهان.31(265):2097-112.
- Hecht JL, Aster JC. Molecular biology of Burkitt’s lymphoma. Journal of Clinical Oncology. 2000;18(21):3707-21.
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98(6):779-90.
- Mohammadi E, Ghaedi K, Esmailie A, Rahgozar S. Gene expression profiling of liver X receptor α and Bcl-2-associated X protein in experimental transection spinal cord-injured rats. The journal of spinal cord medicine. 2013;36(1):66-71.
- Chang T-C, Yu D, Lee Y-S, Wentzel EA, Arking DE, West KM, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nature genetics. 2008;40(1):43-50.
- O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. nature. 2005;435(7043):839-43.
- Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250(4984):1149-51.
- Blackwell T, Huang J, Ma A, Kretzner L, Alt F, Eisenman R, et al. Binding of myc proteins to canonical and noncanonical DNA sequences. Molecular and cellular biology. 1993;13(9):5216-24.
- Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proceedings of the National Academy of Sciences. 1982;79(24):7824-7.
- Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proceedings of the National Academy of Sciences. 1982;79(24):7837-41.
- Ansell SM, Armitage J, editors. Non-Hodgkin lymphoma: diagnosis and treatment. Mayo Clinic Proceedings; 2005: Elsevier.
- Nishikura K, Erikson J, Watt R, Rovera G, Croce C. Differential expression of the translocated and the untranslocated c-myc oncogene in Burkitt lymphoma. Science. 1983;222(4622):390-3.
- Hayday AC, Gillies SD, Saito H, Wood C, Wiman K, Hayward WS, et al. Activation of a translocated human c-myc gene by an enhancer in the immunoglobulin heavy-chain locus. 1984.
- Hann SR, Eisenman RN. Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Molecular and cellular biology. 1984;4(11):2486-97.
- Taub R, Moulding C, Battey J, Murphy W, Vasicek T, Lenoir GM, et al. Activation and somatic mutation of the translocated c-myc gene in Burkitt lymphoma cells. Cell. 1984;36(2):339-48.
- Henglein B, Synovzik H, Groitl P, Bornkamm G, Hartl P, Lipp M. Three breakpoints of variant t (2; 8) translocations in Burkitt’s lymphoma cells fall within a region 140 kilobases distal from c-myc. Molecular and cellular biology. 1989;9(5):2105-13.
- Blomberg BB, Rudin CM, Storb U. Identification and localization of an enhancer for the human lambda L chain Ig gene complex. The Journal of Immunology. 1991;147(7):2354-8.
- Asenbauer H, Klobeck H. Tissue‐specific deoxyribonuclease I‐hypersensitive sites in the vicinity of the immunoglobulin Cλ cluster of man. European journal of immunology. 1996;26(1):142-50.
- Gerbitz A, Mautner J, Geltinger C, Hörtnagel K, Christoph B, Asenbauer H, et al. Deregulation of the proto-oncogene c-myc through t (8; 22) translocation in Burkitt’s lymphoma. Oncogene. 1999;18(9):1745-53.
- Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. 1999;18(19):2942-54.
- Kuttler F, Ame P, Clark H, Haughey C, Mougin C, Cahn J-Y, et al. c-myc box II mutations in Burkitt’s lymphoma-derived alleles reduce cell-transformation activity and lower response to broad apoptotic stimuli. Oncogene. 2001;20(42):6084-94.
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & development. 2000;14(19):2501-14.
- Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin‐mediated proteolysis: cancer‐associated and transforming mutations stabilize Myc. The EMBO journal. 1999;18(3):717-26.
- Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Molecular and cellular biology. 2000;20(7):2423-35.
- Bahram F, von der Lehr N, Cetinkaya C, Larsson L-G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95(6):2104-10.
- پارسا ن. اساس سلولی و مولکولی سرطان در انسان، مقاله مروری. مجله سلول و بافت (Cell & Tissue Journal). 2012;2:365-76.
- Preudhomme C, Dervite I, Wattel E, Vanrumbeke M, Flactif M, Lai JL, et al. Clinical significance of p53 mutations in newly diagnosed Burkitt’s lymphoma and acute lymphoblastic leukemia: a report of 48 cases. Journal of clinical oncology. 1995;13(4):812-20.
- Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, et al. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences. 1991;88(12):5413-7.
- Gaidano G, Hauptschein R, Parsa N, Offit K, Rao P, Lenoir G, et al. Deletions involving two distinct regions of 6q in B-cell non-Hodgkin lymphoma. Blood. 1992;80(7):1781-7.
- Rohn JL, Hueber A-O, McCarthy NJ, Lyon D, Navarro P, Burgering BMT, et al. The opposing roles of the Akt and c-Myc signalling pathways in survival from CD95-mediated apoptosis. Oncogene. 1998;17(22):2811-8.
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, et al. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer cell. 2003;3(5):483-95.
- Kumar A, Marqués M, Carrera AC. Phosphoinositide 3-kinase activation in late G1 is required for c-Myc stabilization and S phase entry. Molecular and cellular biology. 2006;26(23):9116-25.
- Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M. Myc‐induced proliferation and transformation require Akt‐mediated phosphorylation of FoxO proteins. The EMBO journal. 2004;23(14):2830-40.
- Curnock AP, Knox KA. LY294002-Mediated Inhibition of Phosphatidylinositol 3-Kinase Activity Triggers Growth Inhibition and Apoptosis in CD40-Triggered Ramos–Burkitt Lymphoma B Cells. Cellular immunology. 1998;187(2):77-87.
- Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116-20.
- Spender LC, Lucchesi W, Bodelon G, Bilancio A, Karstegl CE, Asano T, et al. Cell target genes of Epstein–Barr virus transcription factor EBNA-2: induction of the p55α regulatory subunit of PI3-kinase and its role in survival of EREB2. 5 cells. Journal of general virology. 2006;87(10):2859-67.
- van Weeren PC, de Bruyn KM, de Vries-Smits AM, Van Lint J, Boudewijn MT. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation characterization of dominant-negative mutant of PKB. Journal of Biological Chemistry. 1998;273(21):13150-6.
- MesrianTanha H, Honardoost MA, Rahgozar S, Ghaedi K. Evaluation of Oncogenic Pathways in T-ALL; New Applications in Treatment. Genetics in the 3rd millennium. 2013;11(2):3100-11.
- Spender LC, Inman GJ. Phosphoinositide 3-kinase/AKT/mTORC1/2 signaling determines sensitivity of Burkitt’s lymphoma cells to BH3 mimetics. Molecular Cancer Research. 2012;10(3):347-59.
- Brennan P, Mehl AM, Jones M, Rowe M. Phosphatidylinositol 3-kinase is essential for the proliferation of lymphoblastoid cells. Oncogene. 2002;21(8):1263-71.
- Devlin JR, Hannan KM, Ng PY, Bywater MJ, Shortt J, Cullinane C, et al. AKT signalling is required for ribosomal RNA synthesis and progression of Eμ‐Myc B‐cell lymphoma in vivo. FEBS Journal. 2013;280(21):5307-16.
- McClure RF, Remstein ED, Macon WR, Dewald GW, Habermann TM, Hoering A, et al. Adult B-cell lymphomas with Burkitt-like morphology are phenotypically and genotypically heterogeneous with aggressive clinical behavior. The American journal of surgical pathology. 2005;29(12):1652-60.
- Dogan A, Bagdi E, Munson P, Isaacson PG. CD10 and BCL-6 expression in paraffin sections of normal lymphoid tissue and B-cell lymphomas. The American journal of surgical pathology. 2000;24(6):846-52.
- Magrath I, Janus C, Edwards B, Spiegel R, Jaffe E, Berard C, et al. An effective therapy for both undifferentiated (including Burkitt’s) lymphomas and lymphoblastic lymphomas in children and young adults. Blood. 1984;63(5):1102-11.
- Kantarjian HM, Walters RS, Keating MJ, Smith TL, O’Brien S, Estey EH, et al. Results of the vincristine, doxorubicin, and dexamethasone regimen in adults with standard-and high-risk acute lymphocytic leukemia. Journal of Clinical Oncology. 1990;8(6):994-1004.
- Bernasconi C, Brusamolino E, Pagnucco G, Bernasconi P, Orlandi E, Lazzarino M. Burkitt’s lymphoma/leukemia: a clinicopathologic study on 24 adult patients. Leukemia. 1990;5:90-4.
- Ostronoff M, Soussain C, Zambon E, Ibrahim A, Bosq J, Bayle C, et al. Burkitt’s lymphoma in adults: a retrospective study of 46 cases. Nouvelle revue française d’hématologie. 1991;34(5):389-97.
- Hoelzer D, Ludwig W, Thiel E, Gassmann W, Löffler H, Fonatsch C, et al. Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood. 1996;87(2):495-508.
- Magrath I, Adde M, Shad A, Venzon D, Seibel N, Gootenberg J, et al. Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. Journal of Clinical Oncology. 1996;14(3):925-34.
- Thomas DA, Cortes J, O’Brien S, Pierce S, Faderl S, Albitar M, et al. Hyper-CVAD program in Burkitt’s-type adult acute lymphoblastic leukemia. Journal of Clinical Oncology. 1999;17(8):2461-.
- Thomas DA, Faderl S, O’Brien S, Bueso‐Ramos C, Cortes J, Garcia‐Manero G, et al. Chemoimmunotherapy with hyper‐CVAD plus rituximab for the treatment of adult Burkitt and Burkitt‐type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106(7):1569-80.
- Ghaedi K. Telomerase: New target for cancer treatment. Genetics in the 3rd millennium. 2009;7(1):1597-603.
- Klapper W, Krams M, Qian W, Janssen D, Parwaresch R. Telomerase activity in B-cell non-Hodgkin lymphomas is regulated by hTERT transcription and correlated with telomere-binding protein expression but uncoupled from proliferation. British journal of cancer. 2003;89(4):713-9.
- Nademanee A, Molina A, O’Donnell MR, Dagis A, Snyder DS, Parker P, et al. Results of high-dose therapy and autologous bone marrow/stem cell transplantation during remission in poor-risk intermediate-and high-grade lymphoma: international index high and high-intermediate risk group. Blood. 1997;90(10):3844-52.
- Song KW, Barnett MJ, Gascoyne RD, Horsman DE, Forrest DL, Hogge DE, et al. Haematopoietic stem cell transplantation as primary therapy of sporadic adult Burkitt lymphoma*. British journal of haematology. 2006;133(6):634-7.
- Serafeim A, Holder MJ, Grafton G, Chamba A, Drayson MT, Luong QT, et al. Selective serotonin reuptake inhibitors directly signal for apoptosis in biopsy-like Burkitt lymphoma cells. Blood. 2003;101(8):3212-9.
- Meredith EJ, Holder MJ, Chamba A, Challa A, Drake-Lee A, Bunce CM, et al. The serotonin transporter (SLC6A4) is present in B-cell clones of diverse malignant origin: probing a potential anti-tumor target for psychotropics. The FASEB journal. 2005;19(9):1187-9.
https://www.cancerresearchuk.org/about-cancer/non-hodgkin-lymphoma/types/burkitt-lymphoma
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام