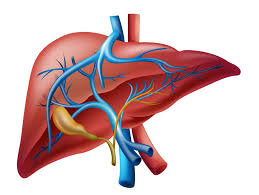
سندرم بود-کیاری
Budd–Chiari syndrome
مهسا جمالی (کارشناس ارشد ژنتیک)
حسن اسدی (کارشناس علوم آزمایشگاهی)

مقدمه
سندرم بود-کیاری بیماری بسیار نادری است که 1 در 1 میلیون نفر از بزرگسالان را تحت تأثیر قرار میدهد. این وضعیت به واسطه انسداد وریدهای کبدی ایجاد میشود. از بارزترین علائم کلاسیک سندرم بود-کیاری، درد شکم، آسیت (آبآوردگی شکم یا تجمع مایع در حفره صفاقی شکم) و بزرگ شدن کبد میباشد. تشکیل لخته خون در وریدهای کبدی میتواند به سندرم بود-کیاری منجر شود. این سندرم میتواند بهصورت برقآسا (خیلی سریع) یا حاد، یا مزمن و یا بدون علامت باشد.

علائم و نشانههای بالینی
سندرم بود-کیاری حاد شامل علائمی مانند درد شدید بالای شکم بهصورت پیشرونده و سریع، تغییر رنگ پوست و سفیدی چشم به زردی، بزرگ شدن کبد، بزرگ شدن طحال، تجمع مایع در حفره صفاقی شکم (آسیت)، افزایش آنزیمهای کبدی و در نهایت آنسفالوپاتی کبدی که باعث ناتوانی کبد در تولید و دفع اوره از آمونیاک میشود، میباشد.

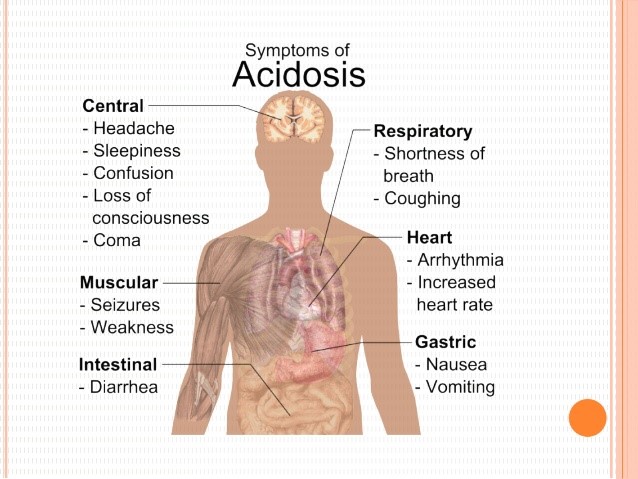
سندرم بود-کیاری برقآسا یا بسیار سریع با علائمی همچون آنسفالوپاتی کبدی و آسیت و همچنین مرگ سلولهای کبدی و اسیدوز لاکتیک (سنتز اسید لاکتیک در جریان خون، زمانی که سطح اکسیژن خون پائین است) شدید همراه است.

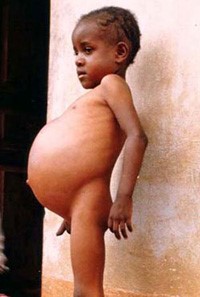
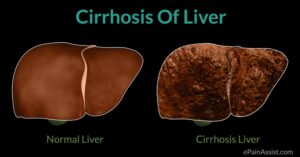
این سندرم در اکثر بیماران مبتلا، بهصورت آهسته و مزمن شروع میشود که این وضعیت میتواند بدون درد باشد. بیماران مبتلا ممکن است به سیروز کبدی دچار شوند و نشانههایی از نارسایی کبدی را بهتدریج نشان دهند. شایان ذکر است که وجود هر یک از علائم ذکرشده به فرم بدون علامت خاص که هیچ نارسایی کبدی همراه با حس درد را به انسان منعکس نکند، دلیلی برای وجود سندرم بود-کیاری نیست.
علتشناسی
در بیشتر از 80% بیماران مبتلا ، علت ایجاد این سندرم شناسایی شده است. سندرم بود-کیاری اولیه در 75% از بیماران مبتلا شامل ترومبوز ورید کبدی است. این در حالی است که خود ترومبوز ورید کبدی با عواملی مانند پلیسایتمی ورا ، بارداری، فشار داخلی پس از زایمان، استفاده بیشازحد قرصهای پیشگیری بارداری، هموگلوبینوری حملهای شبانه (PNH که یک بیماری همولیتیک اکتسابی مزمن است)، سرطان کبد و فاکتور ضدانعقاد لوپوس (Lupus anticoagulant که یک پادتن است و در خون باعث افزایش زمان نسبی ترومبوپلاستین میشود)، همراه است.
در 25% از بیماران مبتلا به سندرم بود-کیاری ثانویه، همانژیوم کبدی مشاهده میشود. اغلب بیماران مبتلا به سمت ترومبوز ورید کبدی گرایش پیدا میکنند که از اولین نشانههای بارز در سندرم بود-کیاری است. نمونههایی از گرایش ژنتیکی سندرم بود-کیاری شامل موارد زیر است:
- کمبود پروتئین C (پروتئین C توسط کبد تولید میشود و کمبود آن روند تنظیم تشکیل لختههای خونی در بدن را دچار اختلال میکند).
- کمبود پروتئین S (کمبود پروتئین s بهصورت اتوزومال غالب به ارث میرسد و کمبود آن به دو شکل هتروزیگوت و هموزیگوت باعث ترومبوز مکرر سیاهرگی و آمبولی ریه میگردد).
- جهش فاکتور انعقادی 5 لیدن (ممکن است شانس لخته شدن خون یا همان ترومبوز را در رگها افزایش دهد).
- کمبود ارثی آنتیترومبین (آنتیترومبین پروتئین تولیدشده توسط کبد میباشد که در جریان انعقاد کمک مهمی به حفظ حالت هوموستاز سیستم انعقادی مینماید).
- جهش ژنتیکی G20210A پروترومبین (جهش G20210A در ناحیه ´3 ژن پروترومبین باعث بالا رفتن میزان این پروتئین در خون میشود که خطر بروز Thrombophilia را به همراه دارد. این ژن نیز بهصورت اتوزومال غالب نسبی به ارث میرسد. جهش G20210A در دو درصد افراد سالم و هفت درصد مبتلایان به ترمبوز وریدی دیده میشود).
یک عامل خطر مهم غیرژنتیکی برای ابتلا ، استفاده از داروهای هورمونی حاوی استروژن بهصورت ترکیبی بهمنظور جلوگیری از بارداری میباشد. از دیگر عوامل خطر برای القاء این سندرم میتوان به سندرم فسفولیپید، آسپرژیلوزیس، بیماری بهجت، مصرف داروهای داکاربازین (داکاربازین برای شیمیدرمانی سرطانهای خونی، لنفوم هوچکین، سرطان پوست، ملانوم بدخیم، سارکوم های بافت نرم و کارسینوم لوزالمعده استفاده میگردد)، بارداری و تروما اشاره کرد.
حدود 12 درصد از بیماران مبتلا که تا 39% توسعه ترومبوز وریدی را نشان میدهند در معرض خطر هموگلوبینوری حملهای شبانه، قرار دارند. همچنین گیرندگان پیوند مغز استخوان جهت درمان انسداد وریدی، ممکن است در معرض سندرم بود-کیاری قرار گیرند.
تشخیص
هنگامی که سندرم بود-کیاری مشکوک است، اندازهگیری میزان آنزیمهای کبدی و مارکرهای اعضای دیگر مانند کراتینین، اوره، الکترولیتها و LDH ضروری است. سندرم بود-کیاری، اغلب با استفاده از مطالعات سونوگرافی از شکم و آنژیوگرافی رتروگراد تشخیص داده میشود. بیوپسی از کبد، غیراختصاصی اما گاهی اوقات لازم است تا این سندرم را از سایر عوامل هپاتومگالی و آسیت مانند گالاکتوزومی کلاسیک و یا سندرم ری، افتراق دهد.
مسیرهای درمانی
اقلیتی از مبتلایان این سندرم را میتوان با داروهای ضدانعقاد مانند هپارین و وارفارین، داروهای دیورتیک یا ادرارآور برای کنترل آسیت در شکم و محدود کردن میزان سدیم در کبد، کنترل و درمان کرد. اکثر مبتلایان سندرم بود-کیاری برای درمان نیاز به مداخله بیشتر تکنیکهای پزشکی دارند. انواع حالتهای خفیف سندرم بود-کیاری را میتوان با جراحی بهمنظور انحراف مسیر جریان خون در اطراف انسداد وریدی یا کبد، کنترل و درمان کرد. بیماران مبتلا به تنگی یا انسداد ورید کاوال که در معرض سندرم بود-کیاری هستند، میتوانند با آنژیوپلاستی نتایج مطلوبی جهت بهبود وضعیت خویش داشته باشند. مطالعات محدودی که در ترومبولیز با تزریق مستقیم اوروکیناز و بافت فعالکننده پلاسمینوژن به داخل ورید مسدود شده، انجام گرفته است، موفقیت نسبی را در درمان سندرم بود-کیاری نشان داده است. بااینحال، این روش بهطور معمول برای همه مبتلایان سندرم بود-کیاری با انسداد وریدی رایج نیست.
پیوند کبد سالم یک مسیر درمانی مؤثر برای سندرم بود-کیاری است. شایان ذکر است که در حدود 10% از بیماران دریافتکننده کبد سالم، سندرم بود-کیاری ممکن است دوباره عود کند.
پیش آگهی
مطالعات متعددی برای بقای مبتلایان این سندرم انجام گرفته است. بهطور کلی 70% بیماران مبتلا به سندرم بود-کیاری تا 10 سال زنده میمانند. شاخص پیشآگهی مهم برای سندرم بود-کیاری شامل آسیت، آنسفالوپاتی کبدی، بالا بودن زمان پروترومبین و متغیر بودن سطوح سرمی از مواد مختلف (سدیم، کراتینین، آلبومین و بیلیروبین) میباشد.
تاریخچه
سندرم بود-کیاری توسط یک پزشک انگلیسی بنام جورج بود و دکتر آرنولد هانس کیاری پاتولوژیست اتریشی در سال 1897 گزارش گردید.


دکتر جورج بود (تصویر سمت راست) و دکتر آرنولد هانس کیاری (تصویر سمت چپ)
References:
- Rajani R، Melin T، Björnsson E، Broomé U، Sangfelt P، Danielsson A، Gustavsson A، Grip O، Svensson H، Lööf L، Wallerstedt S، Almer SH (Feb 2009).
- “Etiology، management، and outcome of the Budd–Chiari syndrome.”
- Hepatic vein thrombosis (Budd–Chiari syndrome).”
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 51-1987. Progressive abdominal distention in a 51-year-old woman with polycythemia vera.”
- “Hepatic outflow obstruction (Budd–Chiari syndrome). Experience with 177 patients and a review of the literature.”
- Podnos YD، Cooke J، Ginther G، Ping J، Chapman D، Newman RS، Imagawa DK (Aug 2003).
- Patel RK، Lea NC، Heneghan MA، et al. (Jun 2006). “Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd–Chiari syndrome”. Gastroenterology. 130 (7): 2031–8.
- Murad SD، Valla DC، de Groen PC، et al. (Feb 2004). “Determinants of survival and the effect of portosystemic shunting in patients with Budd–Chiari syndrome”. Hepatology (Baltimore، Md.). 39 (2): 500–8.
- Budd G (1845). On diseases of the liver. London: John Churchill. p. 135. Brit Lib. 000518193.
- Chiari H (1898). “Erfahrungen über Infarktbildungen in der Leber des Menschen”. Zeitschrift für Heilkunde، Prague. 19: 475–512.
بررسی میزان استرس اکسیداتیو سرم و مایع آسیت بیماران مبتلا به سیروز کبدی
https://medlabnews.ir/%d8%b3%d9%86%d8%af%d8%b1%d9%85-%d8%b3%da%a9%d9%84/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام