مقدمهای بر تکنیک PCR در زمان واقعی و انواع آن
دکتر مهدی فصیحی رامندی (عضو هیئت علمی دانشگاه علوم پزشکی بقیها… (عج))
زهرا کریمی (مرکز تحقیقاتی زیست سلول پژوهان تدبیر)
دکتر رضا میرنژاد (استاد دانشگاه)
PCR در زمان واقعی[1]
آنالیز کمّی توالی اسید نوکلئیک در بسیاری از تحقیقات بیولوژیک نقش مهمی را بازی میکند. اندازهگیری بیان ژن بهطور گسترده در مطالعه پاسخ بیولوژیک به تحریککنندههای سلولی کاربرد دارد، همچنین از این روشها برای تعیین تعداد نسخههای یک ژن در ژنوم موجودات میتوان استفاده کرد؛ بهعنوان مثال ژن Her2 انسانی در 30% از سرطانهای سینه تکثیر میشود. کاربرد دیگر این روشهای کمّی در اندازهگیری میزان ویروس HIV در مراحل مختلف بیماری است. تکنیکهای مختلفی برای مطالعه کمّی توالیهای اسید نوکلئیک وجود دارد. در ابتدا محققین تکنیکهای PCR و RT-PCR کمّی را ارائه کردند. در این تکنیکها میزان محصول PCR در فاز لگاریتمی (قبل از رسیدن به فاز سکون) اندازهگیری میشد. در این روش باید میزان اسید نوکلئیک اولیه همه نمونهها و همچنین کارآیی واکنش PCR در تمام نمونهها یکسان بوده تا نتایج قابل مقایسه باشند. یک ژن مرجع نیز که در همه نمونهها به میزان ثابت و یکسان وجود دارد (مثل β اکتین) برای یکسانسازی واکنش PCR مورد استفاده قرار میگیرد. این روش سخت و تقریباً غیرقابل اعتماد بود، زیرا در این روش باید هم ژن هدف وهم ژن مرجع در فاز لگاریتمی اندازهگیری شوند.
روش دیگر، استفاده از تکنیک PCR کمّی رقابتی[2] است. در این روش از یک کنترل داخلی رقابتی برای یکسانسازی کارایی هر واکنش استفاده میشود. میزان مشخصی از کنترل داخلی رقابتی به هر واکنش اضافه میشود. بهمنظور اندازهگیری نسبی، محصول ژن هدف با محصول ژن کنترل داخلی مقایسه میشود. کلید موفقیت در QC-PCR استفاده از کنترل داخلی مناسب است؛ بهطوریکه کارآیی تکثیر این کنترل داخلی مشابه ژن هدف باشد. طراحی کنترل داخلی و معتبرسازی کارآیی واکنش نیازمند صرف وقت زیادی است، اما از آن جا که در این روش نیازی به بررسی محصول PCR در فاز لگاریتمی نیست، نسبت به روشهای قبلی بهتر است. برای شناسایی و اندازهگیری محصول واکنش در همه روشهای فوق فرایندهای بعد از PCR مورد نیاز است. همین امر موجب صرف هزینه و وقت اضافی شده و میتواند منبع آلودگی باشد، همچنین تعداد نمونههای مورد آزمایش با این روشها محدود است.
روش توسعهیافته دیگر برای آنالیز کمّی DNA طراحی شـــــــده است. این تکنـــــیک که تحت عنوان Real –Time PCR شناخته میشود به مفهوم مشاهده لحظه به لحظه یک فرایند است. این سیستم در سال 1992 کشف و فرمولبندی Real- Time PCR ابتدا توسط Higuchi و همکارانش به انجام رسید.
سایر نامهای این تکنیک عبارتند از quantitative real- time PCR و kinetic PCR لازم به ذکر است که این تکنیک گاهی به اشتباه RT-PCR نامیده میشود.
در این روش از کاوشگرها یا پروبهای هیبریداسیون نشاندارشده با رنگهای فلورسانس در انتهای ‘5 یا ‘3 استفاده میشود که امکان بررسی میزان محصول PCR را بدون جداسازی آنها در روشهای الکتروفورز در ژل آگاروز یا ژل پلیآکریل آمید میدهد. در این تکنیک از یک کاوشگر حاوی دو فلوروکروم استفاده میشود. یک رنگ فلورسنته تحت عنوان گزارشگر[3] FAM (مثل 6-کربوکسی فلورسئین) و رنگ دیگر خاموشکننده[4] مثل TAMRA (مثل 6-کربوکسی تترا متیل رودامین) است. در شرایط عادی به علت نزدیکی دو فلوروکروم به هم هیچ نوری از فلوروکروم گزارشگر ساطع نمیشود، اما هنگامی که آنزیم پلیمراز به کمک فعالیت ‘5 نوکلئازی خود، کاوشگر را تجزیه کند، خاموشکننده از گزارشگر جدا شده و نوری با طولموج 518 نانومتر ساطع میشود. در این سیستم تشخیصی یک مادۀ فلورسنت در طی واکنش متناسب با میزان محصولات هر سیکل آزاد میشود و میزان فلورسنت آن توسط یک نمایانگر3 (مثل ABI Prism) شناسایی و ثبت میگردد. این روش به دلیل کاهش زمان سیکلهای PCR، حذف مرحله Post-PCR و کاربرد نشانگرهای فلوروژنیک و روشهای حساس آشکارسازی تابش آنها باعث افزایش سرعت این سیستم نسبت به سیستم PCR معمولی شده است،اگرچه این روش دارای معایبی نیز هست.
مزایای روش Real- Time PCR
1- تکثیر در هر زمان قابل مشاهده است، به عبارتی امکان مانیتورینگ لحظه به لحظه واکنش فراهم آمده و در هر سیکل امکان بررسی فرآیند تکثیر وجود دارد.
2- مکان بهینهسازی واکنش بهطوریکه مناسبترین غلظت DNA و پرایمر و همچنین تعداد سیکل لازم برای تکثیر مشخص میشود.
3- امکان قطع واکنش در زمان دلخواه: از آنجایی که روند PCR قابل مشاهده است، در صورت عدم تکثیر و یا رفتن به فاز سکون (Plateau)میتوان به واکنش خاتمه داد و از اتلاف وقت و انرژی پرهیز نمود.
4- انجام PCR کمی: با استفاده از روش Absolute Quantification و Relative Quantification میزان دقیق و نسبی الگوی اولیه قابل اندازهگیری است.
5- معمولاً واکنشها سریعتر اتفاق میافتد و زمان کمتری لازم است.
6- محدوده تشخیص آن (Range Detection)بالاتر از PCR معمولی است؛ حتی اختلاف کمتر از 2 برابر را هم نشان میدهد.
کاربردهای Real- Time PCR
- بررسی بیان ژنها: کاربرد وسیع آن در مورد بیان سایتوکاینهای مختلف بخـصوص در مورد بیماریهای خودایمنی و پاتوژنهایی نظیر لیشمانیا است.
- Drug Therapy Efficacy: برای این منظور یک ژن خاص (البته این ژن عامل بیماری نیست و بهعنوان مارکر تلقی میشود) در نظر گرفتـه میشود و اثـر داروی مـوردنظر بـر تغییرات بیان این ژن بررسی میشود. تغییر بیان این ژن نشاندهنده وضعیت درمـانی است.
- بررسی میزان بیان آنزیمهای کبدی برای متابولیز کـردن داروی خـاص: بهطورکلی اثـر داروها بر روی آنزیمهای کبدی به دو صورت است؛ گروهی بیـان آنزیمهای تجزیهکننده داروها را افزایش داده و گروه دیگر باعث مهار آنزیمهای تجزیهکننده داروهـا میشوند. برای داروهای دسته اول مقدار دارو در خون کاهش مییابد و در نتیجـه نیـاز بـه دوز دارویی بالاتری است، ولی در دسته دوم دارو تجزیه نمیشود و در نتیجه مقدار کمتـری دارو موردنیاز است.
- کاربرد در جهت شناسایی و تعیین Load عوامل عفونی: حساسیت بهمراتب بالای این تکنیک امکان تعیین میزان دقیق یک پاتوژن را مهیا کرده است. تعیین میزان دقیق عواملی مثل HIVوHBV در درمان افراد آلوده دارای ارزش است.
- در سیستمهای کنترل غذا و دارو برای Safety Food: نمونه مـوردنظر از نظـر میـزان آلودگی میکروبی با نمونه کنترل مقایسه میشود.
- بررسی سرطانهای خونی و تعیین ریسک بازگشت آن: حساسیت این روش، تشخیص یک سلول سرطانی از میان سلول سالم است که تشخیص قبل، در حین و بعد از درمـان قابـل انجام است.
- در تعیین میزان موفقیت پیوند اعضاء: هر فرد شاخصهای ایمونولوژیک ویژهای دارد. با بررسی این شاخصها در فرد دهنده و گیرنده، قبل و بعد از پیوند و مقایسه آنها باهم میزان موفقیت پیوند مشخص میشود. در صورت موفقیـت پیونـد بایـد شاخصهای فـرد دهنـده در عضو پیوندی پس از پیوند بیشتر باشد.
- ژن درمانی: در این حــالت برای تعیین دوز وکتورحاوی ژن مـــوردنظر در ژن درمــــــــانی یـا Copy Number آن و ارزیابی اینکه وکتور وارد چه سلــــولها یا بافتــــــــی شده اســـت از Real -Time PCR استفاده میشود.
- ژنوتایپینگ: متداولترین کاربرد آن تشخیص قبل از تولد، تـشخیص قبـل از لانـه گزینـی، تعیین subtype ویروسها و باکتریها است.
- تشخیص نـاقلین ناهنجاریهای کرومـوزومی: مثـل سـنــــدرم داون و دوشـــــن کـه در آنDosage Gene تعیین میشود. در این حالت ژنها طوری انتخاب میشوند کـه مقـدار آنها متناسب با کاهش یا افزایش کروموزوم تغییر یابد. معمولاً از ژنهایی استفاده میشود که در هر کرموزوم یک کپی از آن موجود باشد .(Single copy) در صورت تغییر تعداد کروموزومها، تعداد کپی این ژنها نیز تغییر میکند.
- Loss of Hetrozigosity (LOH): در صفتی که رابطه غالب و مغلوبی برقرار باشـد فـرد هتروزیگوت به دلیل داشتن آلل غالب، سالم است. در صورت حـذف یـا جهـش در آلـل غالـب، بیماری در فرد بروز میکند. به این حالت LOH گفته میشود. بـا اسـتفاده از Real- time PCR و از طریق تعیین Copy number ژن موردنظر میتوان به وجود یا عدم وجود این نقص پی برد.
- تشخیص (Single Nucleotide Polymorphism) SNP: جهشهای نقطهای در منطقهای از ژن را میگویند که بهترین مارکرهای اختصاصی ژنتیکی در فرد است که دارای پلیمورفیسم با فراوانی حداقل 1% در جامعه هستند. تشخیص SNP با اسـتفاده از روش Probe FRET قابل انجام است.
انواع رنگهای مورد استفاده در Real- time PCR
هر ماده دارای طولموج تحریک است که این طولموج تنها یک عدد نیست بلکه یک محدوده است؛ یعنی ماده قادر است همۀ طولموجهای این محدوده را دریافت کند، اما جذب ماکزیمم در یک طولموج خاص صورت میگیرد. در مورد طولموج تابش Emission)) هم به همین صورت است. انتخاب رنگها بهمنظور نشاندار کردن پروبها باید طوری باشد که محدوده تابش Reporter (Emission)، با محدوده تحـــریک Quencher (Excitation) همپوشانی داشته باشد (شکل 1). این کار با مقایسه طیف هر رنگ امکانپذیر است، همچنین هنگام انتخاب رنگهای مختلف برای چند پروب در Multiplex Real–Time PCR باید دقت شود که طولموج ساطعشده از هر پروب توسط یک فیلتر خوانده شود.
رنگهای گزارشگر (Reporter Dyes):
|
Lissamine Rhodamine |
FAM[5] | Coumarin | Cy2 | Alexa fluor 633 |
Acridine orange |
|
Cy3.5 |
FITC | Nile Red | Cy3 | Ethidium bromid | Alexa fluor 488 |
| E GFP[6] | JOE | HEX[7] | Cy5 | Nano orange |
Alexa fluor 532 |
رنگهای اطفاکننده (Quencher Dye):
| TAMRA (Tetra Methyl Rhodamine Acronym) |
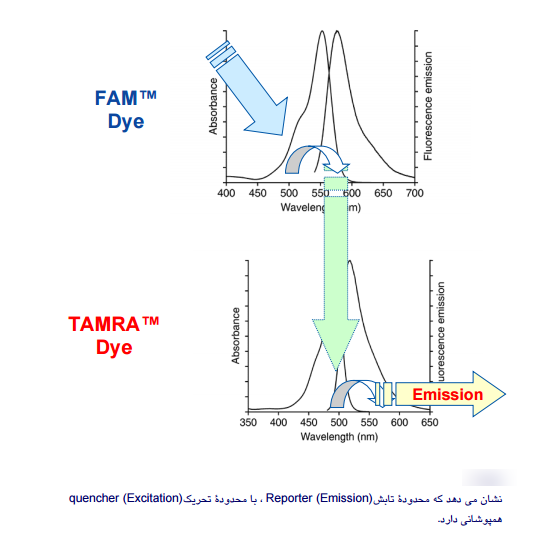
شکل 1: محدوده تابش رنگهای گزارشگر و اطفاکننده
روشهای مختلف Real- Time PCR
بهطورکلی دو روش برای انجام PCR کمی با استفاده از Real -Time PCR داریم:
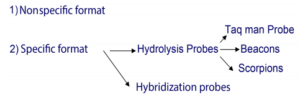
Nonspecific format:
انواع گوناگون رنگهای شیمیایی فلورسنت در فرمهای مختلف در واکنش Real–time PCR استفاده میشود که در سه گروه عمده میتوان آنها را دستهبندی کرد. همه این رنگها میتوانند به مولکول ds DNA به قسمت شیار کوچک مارپیچ DNA متصل شوند و در صورت تحریک با یک طولموج میتوانند در طولموج بالاتری نشر فلورسانس داشته باشند (شکل 2). این رنگها از زمان شناسایی و پیدایش اولین آنها یعنی اتیدیوم بروماید تا به امروز تنوع زیادی پیدا کرده و قابلیتهای بیشتری را کسب کردهاند.
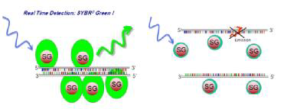
شکل 2: مکانیسم عمل رنگهای فلورسنت
رنگهای گروه یک:
اتیدیوم بروماید تنها عضو این گروه است که خاصیت سمی زیادی برای واکنش PCR دارد، علاوه بر این دارای خاصیت سرطانزایی نیز هست. Higuchi اولین بار از این ماده برای اندازهگیری استفاده کرد. علاوه بر این اتیدیوم بروماید برای رنگآمیزی DNA در ژل آگاروز استفاده میشود.
رنگهای گروه دوم:
این رنگها بهتر از نسل اول بوده و جزو رنگهای Asymmetric cyanin هستند. این رنگها دو حلقه آروماتیک حاوی نیتروژن دارند که یکی از آنها بار مثبت دارد و بهوسیله یک پلیمتیلن به هم متصل شدهاند. این رنگها در حالتی که بهطور آزاد در محلول هستند فاقد فلورسنت هستند، اما بعلت نوسانات و لرزشهایی که هر دو حلقه آروماتیک را شامل میشود انرژی تهییجی الکترونیکی را به حرارت تبدیل میکنند. از انواع این رنگها میتوان به BEBOو SYBER Green I و BOXTO اشاره کرد.
شاید متداولترین رنگ مورد استفاده در این تکنیک سایبرگرین I باشد. این رنگ اینترکاله و فلورسنت، به شیارهای کوچک[8] DNA دو رشتهای متصل میشود و با جذب طولموج 498 نانومتر، نور 522 نانومتری را ساطع میکند که توسط دستگاه ثبت میشود. سایبرگرین به الگوهای تکرشتهای متصل نمیشود، لذا در این موارد بازتاب ضعیفی دارد. در طی چرخههای PCR که محصول دو رشتهای تولید میشود، رنگ به آنها متصل میشود و بنابراین افزایش شدت فلورسنت با غلظت dsDNA متناسب است (شکل3) و در نتیجه، افزایش نور فلورسانس را به همراه دارد. این امر باعث افزایش اتصال رنگ SYBR Green به DNA دو رشتهای شده و افزایش فلورسانس در مرحله پلیمریزه شدن[9] اتفاق میافتد؛ به عبارتی حداکثر فلورسانس در پایان مرحله پلیمریزاسیون و حداقل آن در مرحله واسرشت شدن[10] است. سایبرگرین یک رنگ غیراختصاصی است، لذا برای تمامی آزمایشها قابل استفاده است و این مسئله یک مزیت بهشمار میآید. از دیگر مزایای این رنگ میتوان به ارزانی، عدم تداخل با پلیمرازها، ساده و غیرسمی بودن آن اشاره کرد. همانگونه که گفتیم این رنگ غیراختصاصی است و این مزیت مهمترین عیب آن را نیز شامل میشود چرا که نمیتوان وجود محصولات غیراختصاصی یا پرایمر دایمر را به کمک این رنگ شناسایی کرد. با اتصال به دو رشتهایهایی مثل پرایمر دایمر و دیگر باندهای غیراختصاصی، نتایج بیشتر از غلظت اصلی برآورد میشود و بنابراین اپتیمایزکردن آن باید بهصورتی باشد که حداقل پرایمر دایمر و محصول غیراختصاصی ایجاد گردد. برای تأیید نتایج از آنالیز منحنی ذوب[11] استفاده میشود. از آنجایی که دمای ذوب محصولات PCR بسته به اندازه محصول و توالی نوکلئوتیدهای آن متفاوت است، لذا با استفاده از نمودار دمای ذوب میتوان DNA دو رشتهای هدف را شناسایی کرد. با افزایش تعداد چرخههای PCR، تعداد DNA دو رشتهای نیز افزایش مییابد. فلورسانس ساطعشده بهوسیله دستگاه، قابل شناسایی و اندازهگیری است.

شکل 3: Real -Time PCR با استفاده از رنگ SYBR Green
رنگهای گروه سوم:
شامل رنگهای LC Green وEva Green و CYTO است. این رنگها دارای خاصیت سمی کمتری نسبت به رنگهای نسل اول و دوم برای واکنش PCR هستند. نکته مهم در این رنگها نداشتن خاصـــــــیت ممانعت کنندگی برای آنزیم Polymerase Taq است.
Specific format:
پروبها برای اولین بار در سال 1991 توسط Holland و همکارانش توصیف شدند. پروبها حساسیت و اختصاصیت روش شناسایی را در واکنش Real-time PCR افزایش میدهند، بطوریکه فقط محصول موردنظر شناساییشده و هیچ محصول دیگری قابلشناسایی نخواهد بود.
پروبها برخلاف سایبرگرین بر اساس تشخیص اختصاصی توالی محصول کار میکنند. پروبها معمولاً یکرنگ فلورسنسزا (Reporter) و یکرنگ خاموشکننده (Quencher) دارند. معمولاً دستگاهها بازتابش رنگ فلورسنسزا را بررسی مینمایند. حضور خاموشکننده در نزدیکی موقعیت فلورسنسزا موجب جذب نور و خاموش شدن آن میشود، لذا در این حالت بازتابشی در دستگاه ثبت نمیشود. از این پروبها در روشهای زیر استفاده میگردد:
Hydrolysis probe Technique (1:
این مدل خود به سه دسته قابلتفریق است. در این مدل از مکانیسم FRET[12] استفاده میشود. در این مکانیسم پروبها طوری طراحی میشوند که در ابتدای پروب یک رنگ فلورسانس به نام Reporter و در انتهای آن فلورسانس دیگری به نام Quencher قرار میگیرد. وقتیکه Reporter و Quencherدر فاصلۀ مولکولی نزدیک قرار دارند (در حالت اتصال به پروب) نوری که به Reporter میخورد باعث ایجاد یک Emission میشود که طولموج این Emission در ناحیۀ تحریک Quencher است و Quencher این نور را جذب کرده وEmission ای در طولموج بلندتر ساطع میکند که قابل ارزیابی توسط دستگاه نیست. پس از جدایی Quencher و Reporter در این حالت نور ساطعشده از Reporter توسط Quencher قابلجذب نیست و توسط دستگاه بهصورت فلورسانس قابلاندازهگیری است.
Taq Man Probe:
نمونه بارز این روش استفاده از کاوشگرهایی مرسوم به کاوشگر[13] TaqMan یا کاوشگرهای اولیگونوکلئوتیدی دارای دو رنگ[14] است. اساس آن خاصیت ´5 Exonuclease Activity آنزیم Taq DNA Polymerase است. آنزیم Taq DNA Polymerase با استفاده از خاصیت اگزونوکلئازی خود پروب را تجزیه میکند که این خاصیت اساس کار این روش است (شکل4).
کاوشگر در انتهای ´5 توسط ماده فلورسنت گزارشگر و در انتهای ´3 توسط ماده فلورسنت خاموشکننده نشاندار میشود. در این روش از خاصیت اگزونوکلئازی ´3 → ´5 آنزیم Taq پلیمراز استفاده میشود. اساس کار به این شکل است که تا هنگامی که کاوشگر سالم و دستنخورده است، به دلیل نزدیکی گزارشگر به خاموشکننده، از ساطع شدن فلورسانس فلوروکروم گزارشگر جلوگیری میشود. هنگام تکثیر توالی هدف و در مرحله پلیمریزاسیون، کاوشگر بهوسیله آنزیم Taq پلیمراز از DNA جدا شده و بهواسطه فعالیت اگزونوکلئازی´3 → ´5 آنزیم Taq پلیمراز، هیدرولیز میشود. با هیدرولیز کاوشگر، فلوروکروم گزارشگر و خاموشکننده از یکدیگر جدا میشوند. جدایی دو فلوروکروم از هم موجب میشود فلورسانس گزارشگر، ساطع و در نتیجه قابل شناسایی میشود. پس از اتمام هر چرخه PCR، مولکولهای بیشتری از فلوروکروم گزارشگر جدا شده و میزان فلورسانس پس از انجام هر چرخه موفق PCR، افزایش مییابد. فلورسانس ساطعشده بهوسیله دستگاه قابل شناسایی و اندازهگیری است.
دو نوع خاموشکننده وجود دارد:
- خاموشکننده گرمایی: این خاموشکننده زمانی کار میکند که فاصله آن با فلوروفور کمتر از 10 آنگستروم باشد. این خاموشکننده انرژی را بهصورت نور دریافت کرده و به گرما تبدیل میکند.
- خاموشکننده نوری: که در فاصله 100-10 آنگسترومی از فلوروفور عمل کرده و نور دریافتی را با طولموج بالاتر تابش میکند. فاصله بین فلوروفور و خاموشکننده نوری در حدود 40 باز است.
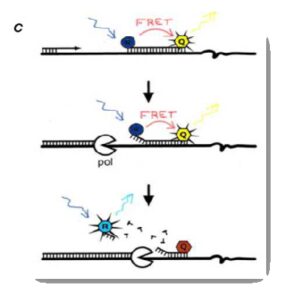
شکل 4: Real- Time PCR با استفاده از کاوشگر هیدرولیزی
Molecular Beacons:
این پروب نیز همانند پروب Taq Man دارای رنگ در انتهای́ 3 و ‘5 است، اما برخلاف Taq Man تجزیه نمیشود و در سیکلهای بعدی دوباره استفاده میگردد. این پروب قبل از اتصال به DNA بهصورت لوپ است که به آن Structure Stem loop میگویند.
بیکونهای مولکولی (Molecular beacons) سادهترین پروبهای سنجاقسری هستند که شامل یک ناحیه با ترادف اختصاصی (ناحیه لوپ) میباشند که دو طرف آن توالیهای معکوس تکراری وجود دارد. رنگهای Reporter و Quencher به دو انتهای مولکول متصل میشوند.
به علت نزدیکیReporter و Quencher نور ساطعشده توسط Quencher مهار میشود. پس از اتصال پروب به محل موردنظر خود، ساختار Stem loop باز شده و در نتیجه رنگها از هم دور میشوند و تابش نور از Reporter انجام میگیرد (شکل 5).

شکل 5: ساختار عملکرد بیکونهای مولکولی
Scorpion Primer:
شکل آزاد پروبهای اِسکورپیون با بیکون مولکولی در محلول شباهت دارد و ساختار سنجاقسری تشکیل میدهند، با این تفاوت که یکی از پرایمرهای تکثیری با اتصال کوالانت به توالی پروب متصل شده است. برخلاف بیکونهای مولکولی، ساختار سنجاقسری اِسکورپیون به انتهای ‘5 پرایمر اختصاصی متصل میشود.
توالی پروب طوری طراحی شده که مکمل تعدادی از نوکلئوتیدها بعد از پرایمر Forward است. دو طرف پروب شامل توالی شش نوکلئوتیدی است که در دمای محیط ایجاد Duplex میکند. انتهای ‘5 ساقه بهصورت کووالان به Reporter و انتهای ‘3 به Quencher متصل است. هنگامی که پروب بهصورت Duplex است، نور ساطع نمیشود. یک مسدودکننده نیز از تکثیر پروب توسط پرایمر Reverse در چرخه بعدی PCR جلوگیری میکند. جهتگیری پروب به صورتی است که انتهای ‘5 آن مکمل انتهای ‘3 قطعه هدف است. در طول سیکلهای PCR، همزمان با اتصال پرایمرها، لوپ پروب باز شده و به قسمت تکثیرشده از چرخۀ قبل متصل میشود و در نتیجه به علت جدا شدن Reporter از Quencher نور ساطع میشود. در مرحلۀ بعد پروب از Template جدا شده و به حالت خاموش درمیآید. به این ترتیب در هر Annealing با افزایش محصول PCR مقدار فلورسانس نیز افزایش مییابد.
Hybridization probes (2:
در این روش از دو کاوشگر اختصاصی مربوط به توالی هدف استفاده میشود. هر دو کاوشگر با ماده فلورسنت نشاندار شدهاند (کاوشگر اول در انتهای ´3 با فلوروکروم دهنده مثلاً FAM و کاوشگر دوم در انتهای ´5 با فلوروکروم گیرنده مثلاً 705 یا LC Red 640 نشاندار میشوند). کاوشگر فرادست دهنده و کاوشگر پائیندست گیرنده است. کاوشگرها به توالیهای مکمل خود در DNA تکثیرشده، متصل میشوند. اتصال کاوشگرها به توالی هدف در DNA تکثیرشده، سبب میشود که دو کاوشگر به فاصله 5-1 نوکلئوتیدی از هم قرار بگیرند (شکل 6).
این امر موجب میشود فلورکروم دهنده برانگیخته شده و نور را با طولموج بلندتر ساطع کند. بهواسطه انتقال انرژی رزونانس فلورسنس (FRET)[15]، فلوروکروم پذیرنده نیز برانگیخته شده و فلورسانس قابل شناسایی برای دستگاه ساطع میشود. فلورسانس ساطعشده در مرحلة اتصال کاوشگر و ابتدای مرحلة پلیمریزاسیون شناسایی میشود. با افزایش تعداد چرخههای موفق PCR، میزان اتصال کاوشگر افزایش مییابد که این امر به افزایش فلورسانس ساطعشده منجر میگردد. در این روش، هنگامی که کاوشگرها به الگو متصل نشدهاند، کاوشگر دهنده از خود نور ساطع میکند؛ لذا باید فیلتری در دستگاه تعبیه شود تا این نور شناسایی نشود. از آنجا که در این روش از دو کاوشگر استفاده میشود، اختصاصیت آزمایش افزایش مییابد. پلیمراز مورد استفاده در این روش نباید خاصیت نوکلئازی داشته باشد، زیرا در این صورت موجب تجزیه و تخریب کاوشگر میشود. طراحی کاوشگر و بهینهسازی آزمایش مشکل است.

شکل 6: Real- Time PCR با استفاده از کاوشگر هیبریداسیونی
مفاهیم مهم در Real- Time PCR
(Threshold) Crossing Line
بر اساس شدت فلورسانس زمینه[16] معمولاً بین چرخه 15-3 آستانهای برای فلورسانس اختصاصی تعیین میشود که از این Cut off بهعنوان Crossing Line نام برده میشود.
Crossing point= (Cp) Or (Cycle threshold) = (Ct)
چرخهای از PCR که فلورسانس اختصاصی برای اولین بار از Crossing line میگذرد را Ct یا Cp گویند. در واقع هرچه Crossing Point پایینتر باشد تعداد نسخه[17] هدف بیشتر است.
منحنی استاندارد[18]
برای محاسبة کمی تعداد کپی ژن هدف، از منحنی استاندارد استفاده میشود. بدین منظور رقتهای متوالی از ژن موردنظر را تهیـــه کرده و بــــر روی رقتهای تهیهشده، Real Time PCR انجام میشود. با انجام Real -Time PCR مقادیر Crossing Point رقتهای مختلف بهدست میآید.
با استفاده از دو فاکتور رقت و Crossing Point نمودار استاندارد تهیه میشــــــــود. بــــــــا انجام Real -Time PCR برای نمونههای مجهول و بهدست آوردن Crossing Pointهای آنها میتوان رقت نمونة مجهول را با استفاده از منحنی استاندارد محاسبه کرد. عمـــلاً هرچـــه غلظت بیشـــتر باشد، Crossing Point پایینتر است.
انواع اندازهگیریها در تکنیک Real -Time PCR
یکی از مهمترین کاربردهای Real -Time PCR تعیین کمی مقدار ژن بیانشده است که دو روش عمده برای بررسی کمی در Real- Time PCR وجود دارد:
- Absolute Quantification
- Relative Quantification
در Absolute Quantification تعداد دقیق کپی از یک ژن (Copy number) مشخص میگردد درحالیکه Relative Quantification ، نسبت بیان یک ژن به ژن دیگر یا به عبارتی تغییرات کمی در بیان یک ژن اعلام میشود.
Absolute Quantification (مقایسه مطلق) (منحنی استاندارد):
برای تعیین دقیق کپیهای یک ژن (Copy Number) به منحنی استاندارد نیاز است.
در این روش از نمونۀ RNA یا DNA با غلظت مشخص برای رسم منحنی استاندارد استفاده میشود. غلظت RNA یا DNA استاندارد با اسپکتروفتومتر (در طولموج nm260) تعیین میشود و سپس از روی وزن مولکولی نمونه به تعداد نسخههای آن تبدیل میگردد. لازم به ذکر است که استانداردهای غلظتی ژنهای معروف بهصورت تجاری قابل خریداری است، هرچند که بسیار گرانقیمتاند.
برای حذف نوسانات مقادیر RNAواردشده در واکنش و خطاهای عملکرد دستگاهها و فرد از استانداردهای داخلی استفاده میشود. این استانداردها باید در همۀ بافتها بیان ثابت داشته باشند و آزمایش ما نباید بیان آن را نسبت به نمونۀ کنترل تغییر دهد؛ بدین منظور از ژن بتا اکتین (Beta actin) و Glyceraldehyde-3-Phosphate dehydrogenase (GAPDH) استفاده میشود. به این ژنها Housekeeping میگویند. علاوه بر برپایی PCR با پرایمر اختصاصی ژن، برای هر نمونه یک PCR همزمان با پرایمر بتااکتین یا GAPDH هم انجام میشود و دادههای هر نمونه با ژن Housekeeping همان نمونه نرمالایز میشود، سپس با استفاده از نمودار استاندارد میتوان به غلظت نمونۀ مجهول پی برد.
منحنی استاندارد بر اساس مقدار معلوم همان ژن و Cycle Threshold(چرخه آستانه) هر نمونه رسم میشود. در ادامه یک سری اصطلاحات رایج در Real-Time PCR توضیح داده میشود.
اصطلاحات متداول در Time PCR–Real:
| Amplification plate | یا نمودار تکثیر، نموداری است که در آن تعداد چرخهها در مقابل شدت نور فلورسنت رسم میشود. منحنی تکثیر تغییرات فلورسانس را روی محور y نشان میدهند و محور افقی تعداد سیکل است. |
|
Base line
|
خط پایه، به چرخههای ابتدایی گویند که در آنها شدت نور فلورسنت تغییر معنیداری نکرده است که معمولاً بین 3 تا 15 سیکل اول است. |
| Number CT | چرخۀ آستانه، به اولین چرخههایی گویند که شدت نور فلورسنت در آنها بهصورت معنیداری بیشتر از خط پایه است. |
|
Normalization
|
نرمالسازی، عبارت است از بکار بردن یک استاندارد بیاثر و بدون تغییر در واکنش تا به کمک آن بتوان خطاهای مراحل آزمایش را کم نمود یا از بین برد. |
| Passive reference | رفرانس بیاثرROX ، یک رنگ برای نرمالسازی بهشمار میآید. معمولاً از رنگ برای حذف نوسانات تابش و دریافت نور در لولههای متفاوت PCR استفاده میشود، درواقع ROX رنگی Reference dye است که باعث تصحیح نوسانات فلورسانس میگردد و در ایجاد سطحی از فلورسانس جهت تعیینBackground بهکار میرود. |
| Standard curve | منحنی استاندارد، تعداد CT در مقابل لگاریتم غلظت یک نمونه استاندارد را نمایش میدهد و از این نمودار برای تخمین غلظت نمونههای مجهول استفاده میشود. |
| Threshold | آستانه، خطی است که منحنی تکثیر را در فاز لگاریتمی قطع میکند. |
| ΔRn | اختلاف بین میزان بالاترین فلورسانس با پایینترین میزان فلورسانس است. نمونههای مثبت باید ΔRn مثبت داشته باشند. |
| Slope | شیب یا انحراف، کارایی و راندمان منحنی Time PCR –Real را نشان میدهد، بطوریکه در یک منحنی خوب وقتی آستانه و خط پایه را تنظیم میکنیم، شیبی معادل 3/23- داشته باشد، بدین معنی که (3/2- تا 3/6-) بهترین محدوده شیب است. |
| CT | محل تلاقي منحني استاندارد با خط Threshold یا آستانه را میگویند. |
| NTC | کنترل منفی که فاقد ژن الگو (Template) است. |
Relative Quantification (مقایسه نسبی):
یکی از کاربردیترین استفادههای Real-Time PCR بررسی بیان ژنهاست که با استفاده از کمیتسنجی نسبی (Relative Quantification) انجام میشود. در حال حاضر Relative Quantification دقیقترین روش برای بررسی تغییرات بیان ژن است. برای حذف نوسانات مقادیر RNA واردشده در واکنش و خطاهای عملکرد دستگاهها و فرد از استانداردهای داخلی استفاده میشود. این استانداردها باید در همۀ بافتها بیان ثابت داشته باشند و آزمایش ما نباید بیان آن را نسبت به نمونۀ کنترل تغییر دهد. در این روش تعداد مهم نیست و فقط کاهش یا افزایش بیان ژن مهم است که این کاهش یا افزایش را با یک استاندارد یا ژن reference مقایسه میکنند. همانطور که قبلاً اشاره شد این استاندارد بهعنوان کنترل داخلی در نظر گرفته میشود. این ژن عموماً یک Gene House keeping است.
ویژگیهای ژن استاندارد عبارتند از:
- در همه سلولها Copy number یکسانی داشته باشد.
- در همه سلولها بیان شود.
- بیان ژن مرجع و ژن موردنظر باید متناسب باهم باشند. اگرژن موردنظر به میزان کم بیان میشود نباید ژن مرجعی را انتخاب کنیم که بیان آن زیاد است، در این صورت مقایسه یک مقدار کم با مقدار زیاد باعث افزایش خطای کار میشود.
- از تیمارگریهای واکنش نباید تأثیر بپذیرد.
استانداردهایی که بهطور عموم استفاده میشوند عبارتند از:
1- β-actin mRNA
2- MHC-I mRNA (Major Histocompatibility Complex)
3- Cyclophilin mRNA
4- mRNA for certain ribosomal proteins
5- 28 S or 18 S rRNA
6- Glyceraldehyde3-phosphate dehydrogenase mRNA
تحقیقات نشان داده است که بعضی از این ژنها در شرایط آزمایشگاه تغییر میکنند. نکته مهم این است که این ژن، در حین آزمایش موردنظر مقدار ثابتی داشته باشد. ژنهای کنترل را میتوان همزمان بهصورت Multiplex با ژن موردنظر در یک Tube تکثیر کرد که در این صورت باید از پروب استفاده شود و یا اینکه بهصورت جدا در Tube دیگر تکثیر گردد.
در Relative Quantification لزومی به دانستن غلظت دقیق نمونهها نیست. این روش هم از نظر اقتصادی و هم از نظر زمانی مقرون بهصرفه است. در این روش میتوان از cDNA ,RNA، DNA ژنومی و یا حتی DNA کلونشده استفاده کرد.
اساس روش Relative Quantification برپایه نسبت بیان ژن موردنظر به ژن مرجع است. باید دقت شود که PCR Efficiencyهر دو ژن تقریباً برابر باشد، در غیر این صورت استفاده از این روش خطای زیادی ایجاد میکند.
پردازش اطلاعات در Real–time PCR بسیار حائز اهمیت است. پردازش بر اساس نمودار استاندارد و ارزیابی بازده PCR انجام میگیرد. درRelative Quantification ، مهم بازده PCR است، درصورتیکه در Absolute Quantification نمودار استاندارد حائز اهمیت است. برای بررسی و محاسبه بیان ژن موردنظر و ژن مرجع به روشهای ریاضی متعددی نیاز هست.
برای بهدست آوردن نسبت ژن موردنظر به ژن مرجع میتوان از طریق فرمول زیر محاسبه کرد و اساس آن بر پایه Efficiency و اختلاف در Ct اسـت:

Control = قبل از Treatment
Sample= بعد از Treatment
اگر بازده PCR را کامل در نظر بگیریم فرمول بهصورت زیر تبدیل میشود:

نرمافزارهای مختلفی برای بررسی اختلاف بین بیان ژن نمونه و مرجع وجود دارد از جمله:
.Q-Gene, Light Cycler Quantification Software
در صورتی که تعداد نمونهها زیاد باشد (تا 100 نمونه) میتوان از نرمافزارهای REST و REST- XL استفاده کرد.
در برنامه Q-Gene که یک برنامه مقایسهای Excel است، پردازش ورودیها، آنالیز دادهها و رســــم نمودارها انجام میشود. Q-Gene چندین برنامه آماری دارد و نرمالایزکردن بیان ژن را انجام میدهد.
یک نمونه از پروتکل Real- Time PCR با روش مقایسه مطلق
مواد موردنیاز:
لیست مواد مورد استفاده و غلظت مناسب برای انجام آزمایش Real -Time PCR در جدول زیر آمده است.
جدول 1: مواد مورد استفاده و غلظت مناسب برای انجام آزمایش Real- Time PCR
| غلظت نهایی | حجم | مواد |
| 11μl | PCR-grade water | |
| 1X | 2/5μl | 10 X PCR buffer |
| 8 mM | 4μl | Mgcl2(50 mM) |
| 1 X | 5μl | 5 X reagent mix (containing dNTP,primers and probe) |
| 1/5 واحد | .0/5μl | Taq Polymerase |
| 2μl | cDNA | |
| 25μl | Total Volume |
روش کار:
1- ابتدا از مواد موردنیاز برای آزمایش Real- Time RT-PCR (بجز Diluted cDNA) یک مخلوط اصلی[19] تهیه و به مقدار 15 میکرولیتر از این مخلوط را در هر میکروتیوب 0/1 میلیلیتری بریزید. معمولاً داخل کیت، میکروتیوبهای حاوی تعداد مشخصی از نسخههای ژن هدف بهصورت coatشده[20] نیز وجود دارد، در نتیجه نیازی به افزودن cDNA نیست و میبایست 10 میکرولیتر آب تیمارشده با DEPC به آن افزود تا حل شود (میکروتیوب استاندارد)، اما به سایر میکروتیوبها میبایست علاوه بر 15 میکرولیتر از مخلوط اصلی، 2 میکرولیتر cDNA نمونه افزوده گردد.
در مورد نمونه کنترل منفی (NTC)[21]، میبایست بهجای cDNA بیمار، آب اضافه گردد تا هیچ الگویی برای انجام واکنش PCR در آن وجود نداشته باشد.
2- درب میکروتیوبها را بسته و درون دستگاه قرار دهید. سپس یک برنامه زمانی دو مرحلهای، طبق جدول زیر برای دستگاه تعریف نمایید.
جدول 2: برنامه زمانی جهت اجرای Real- Time PCR
| زمان | دما | |
| 2min | 95°c | HOLD |
| 15sec | 95°c | Cycles |
| 1min | 95°c | |
| 40°c | Cycles Number | |
طبق این برنامه زمانی، ابتدا دمای 95 درجه سلسیوس به مدت دو دقیقه جهت واسرشت شدن اولیه نمونه اعمال میشود. سپس در هر چرخه، ابتدا دمای 95 درجه سلسیوس به مدت 15 ثانیه جهت واسرشت شدن و سپس دمای 59 درجه سلسیوس به مدت یک دقیقه جهت اتصال پرایمرها و کاوشگر به نمونه و فعالیت آنزیم پلیمراز اعمال خواهد گردید. از آنجا که دو مرحله اتصال پرایمرها و فعالیت آنزیم پلیمراز با هم ادغام و طی یک مرحله با دمای مشترک 59 درجه سلسیوس صورت میگیرد، به این برنامه، برنامه دو مرحلهای[22] برای PCR میگویند. در واقع بهجای حضور سه مرحله[23] اصلی در برنامه PCR یعنی مرحله واسرشت شدن، اتصال پرایمرها و کاوشگر و فعالیت آنزیم، بهطور جداگانه در این برنامه، دو مرحله وجود دارد.
علت انتخاب این برنامه، مناسب بودن دمای 59 درجه سلسیوس برای فعالیت اگزونوکلئازی آنزیم در راستای ایجاد نور فلوئورسانس است که در این دما پرایمرها و کاوشگر نیز متصل میگردند.
آزمایش Real- Time RT-PCR برای ژن مرجع:
تمام مواد موردنیاز برای انجام این آزمایش، روش کار و برنامه زمانیPCR آن کاملاً مشــــابه با آزمایش Real- Time PCR برای ژن مورد بررسی است، با این تفاوت که میبایست از پرایمرها و کاوشگر و میکروتیوبهای استاندارد مخصوص ژن مرجع استفاده نمود.
محاسبه تعداد نسخههای m-RNA ژن مرجع و هدف در نمونهها
پس از اتمام 40 چرخه، اطلاعات بهدستآمده در کامپیوتر متصل به دستگاه، ذخیره و با استفاده از میکروتیوبهای استاندارد و تعیین تعداد دقیق نسخههای m-RNA هر دو ژن، منحنی استاندارد رسم میشود. در واقع، دستگاه با رسم منحنی استاندارد هر ژن، بهطور اتوماتیک تعداد نسخههای m-RNA آن ژن را محاسبه و اعلام مینماید.
[1] Real- Time PCR
[2] Quantitative competitive (QC)-PCR
[3]– Reporter
[4] -Quencher
[5] 6-carboxy-fluorescein
[6] Green Fluorescent Protein
[7] Hexa chlorfluorescein
[8] -Minor groove
[9] -Extension
[10]– Denaturation
[11] – Melting Curve
[12] Forster Transfer Probes
[13]-Probe
[14] -Double dye oligonucleotide probe
[15] -Fluorescence Resonance Energy Transfer
[16] – Background
[17] – Copy number
[18]– Standard curve
[19] – Master Mix
[20] – Coated cDNA
[21] – Non Target Control
[22] -Two Step PCR
[23] -Three Step PCR
روشهای عملی در Real Time- PCR
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام