سندرم رحمان
Rahman Syndrome
شاهین اسعدی (دانشجوی دکتری تخصصی ژنتیک پزشکی)

کلیاتی از سندرم رحمان
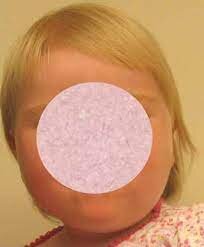
سندرم رحمان یک اختلال ژنتیکی نادر است که با ناتوانی ذهنی خفیف تا شدید با رشد بیش از حد متغیر بدنی همراه با افزایش طول قد، وزن و یا اندازه دور سر (ماکروسفالی) مشخص میشود.

شکل 1: تصاویری از مبتلایان سندرم رحمان همراه با اختلال ماکروسفالی
علائم و نشانههای بالینی سندرم رحمان
در این سندرم، رشد بیش از حد در نوزادی آشکار است و ممکن است با گذشت زمان کاهش یابد یا ادامه داشته باشد. فنوتیپ مبتلایان این سندرم، بسیار متغیر است. برخی از افراد مبتلا، ممکن است ناهنجاریهای جزئی دیگری داشته باشند، از جمله ویژگیهای بدشکل صورت، استرابیسم (لوچی چشم) یا کامپتوداکتیلی (ناهنجاری انگشتان). تصور میشود که این اختلال ناشی از نقص در تنظیم اپیژنتیک باشد.
علاوه بر موارد ذکرشده، علائم دیگر سندرم رحمان شامل کاهش تون عضلانی در دوره نوزادی (هایپوتونی) و در بعضی موارد نادر، افزایش تون عضلانی (هایپرتونی)، گونههای کامل، خط موی بالا و تلکانتوس، کیفواسکولیوز، کامپتوداکتیلی (خمیدگی انگشتان)، تالیپس اکوینوواروس، سن پیشرونده استخوان، ناهنجاریهای دندانی، خالهای پوستی، استرابیسم، آستیگماتیسم و آمبلیوپی است.
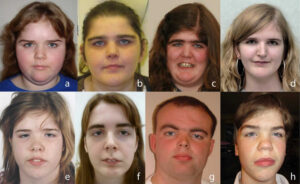
شکل 2: نمایی دیگر از مبتلایان سندرم رحمان
علتشناسی سندرم رحمان
محققان تاکنون در 5 بیمار غیر مرتبط با سندرم رحمان، 3 جهش کوتاهکننده مختلف هتروزیگوت در ژن HIST1H1E که در بازوی کوتاه کروموزوم شماره 6 به صورت 6p22.2 مستقر است، شناسایی کردهاند. جهشهایی که با تعیین توالی تمام اِگزوم پیدا شدهاند و با توالی سنگر تأیید شدهاند، در 4 خانواده از نوع جهشهای جدید بودند. DNA والدین از خانواده پنجم در دسترس نبود. همه جهشها منجر به تولید پروتئین مشابه کوتاهشده در حوزه C-terminal میشوند که در اتصال کروماتین و فعل و انفعالات پروتئین-پروتئین نقش دارد. پیشبینی میشود که پروتئینهای کوتاهشده در مقایسه با پروتئین نوع وحشی (نرمال) بار خالص کاهشیافته داشته باشد و این امر باعث میشود که در خنثیسازی DNA اتصالدهنده با بار منفی، کارآیی کمتری داشته باشند. علاوه بر این، کوتاه شدن انتهای C احتمالاً مانع فعل و انفعالات DNA و پروتئین-پروتئین میشود.
سندرم رحمان در برخی موارد از الگوی توارثی اتوزومال غالب پیروی میکند؛ بنابراین برای ایجاد این سندرم، یک نسخه از ژن جهشیافته HIST1H1E (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزند مبتلا به این سندرم در حالت اتوزومی غالب، برای هر بارداری احتمالی به میزان 50% است. لازم به ذکر است که در بیشتر موارد، سندرم رحمان در اثر جهشهای جدید (de novo) و بدون سابقه خانوادگی رخ میدهد.
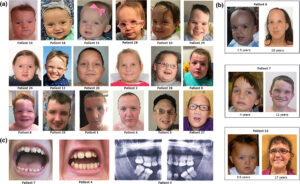
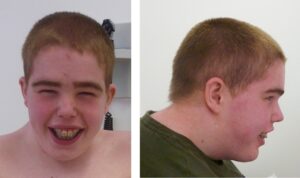
شکل 3: تصاویری از مبتلایان سندرم رحمان همراه با اختلالات مربوطه
فراوانی سندرم رحمان
سندرم رحمان اختلال ژنتیکی بسیار نادری است که فرکانس دقیق شیوع آن در جهان مشخص نیست.
![]()
شکل 4: شماتیکی از کروموزوم شماره 6 که ژن HIST1H1E در بازوی کوتاه این کروموزوم به صورت 6p22.2 مستقر است
تشخیص سندرم رحمان
سندرم رحمان بر اساس یافتههای بالینی و فیزیکی مبتلایان و برخی آزمایشهای پاتولوژیکی، قابل تشخیص است. دقیقترین روش تشخیص این سندرم، آزمایش ژنتیک مولکولی برای ژن HIST1H1E به منظور بررسی وجود جهشهای احتمالی است.
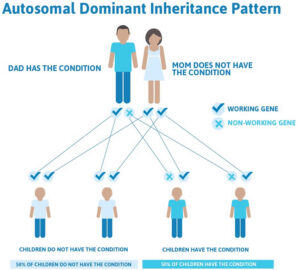
شکل 5: شماتیکی از الگوی توارثی اتوزومال غالب که سندرم رحمان از این الگو تبعیت میکند
مسیرهای درمانی سندرم رحمان
استراتژی درمان و مدیریت سندرم رحمان به صورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص مغز و اعصاب، متخصص طب فیزیکی، متخصص ارتوپدی، جراحان و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان مؤثری برای این سندرم وجود ندارد و تمامی اقدامات بالینی به منظور تخفیف رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند، ضرورت دارد.
منبع:
اسعدی شاهین، کتاب پاتولوژی در ژنتیک پزشکی جلد 18 (A-H)، صفحات 937-932، انتشارات کتب دانشگاهی عمیدی، بهار 1400.
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام